UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the Fiscal Year Ended December 31, 2011
Commission File Number 333-150149
TONIX PHARMACEUTICALS HOLDING CORP.
(Exact name of registrant as specified in its charter)
| Nevada | 26-1434750 | |
|
(State or other jurisdiction of incorporation or organization) |
(IRS Employer Identification No.) | |
|
509 Madison Avenue, Suite 306 New York, New York |
10022 |
(212) 980-9155 |
| (Address of principal executive office) | (Zip Code) | (Registrant’s telephone number, Including area code) |
Securities registered pursuant to Section 12(b) of the Act: None
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined by Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 229.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ¨ | Accelerated filer ¨ |
| Non-accelerated filer ¨ | Smaller reporting company x |
| (Do not check if a smaller reporting company) |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).Yes¨ Nox
There was no aggregate market value of the voting common stock held by non-affiliates as of June 30, 2011, as our common stock was not publicly traded at that time.
As of March 20, 2012, there were 34,278,432 shares of registrant’s common stock outstanding.
TABLE OF CONTENTS
| PAGE | |||||
| PART I | |||||
| Item 1. | Business | 3 | |||
| Item 1A. | Risk Factors | 21 | |||
| Item 1B. | Unresolved Staff Comments | 37 | |||
| Item 2. | Properties | 37 | |||
| Item 3. | Legal Proceedings | 37 | |||
| Item 4. | Mine Safety Disclosures | 37 | |||
| PART II | |||||
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | 38 | |||
| Item 6. | Selected Financial Data | 39 | |||
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations | 39 | |||
| Item 7A. | Quantitative and Qualitative Disclosures about Market Risk | 45 | |||
| Item 8. | Financial Statements and Supplementary Data | F1-F17 | |||
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosures | 46 | |||
| Item 9A. | Controls and Procedures | 46 | |||
| Item 9B. | Other Information | 47 | |||
| PART III | |||||
| Item 10. | Directors, Executive Officers and Corporate Governance | 48 | |||
| Item 11. | Executive Compensation | 52 | |||
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 54 | |||
| Item 13. | Certain Relationships and Related Transactions, and Director Independence | 56 | |||
| Item 14. | Principal Accounting Fees and Services | 56 | |||
| PART IV | |||||
| Item 15. | Exhibits | 57 | |||
| Signatures | 60 | ||||
| 2 |
PART I
ITEM 1 - BUSINESS
This Annual Report on Form 10-K (including the section regarding Management's Discussion and Analysis of Financial Condition and Results of Operations) contains forward-looking statements regarding our business, financial condition, results of operations and prospects. Words such as “expects,” “anticipates,” “intends,” “plans,” “believes,” “seeks,” “estimates” and similar expressions or variations of such words are intended to identify forward-looking statements, but are not deemed to represent an all-inclusive means of identifying forward-looking statements as denoted in this Annual Report on Form 10-K. Additionally, statements concerning future matters are forward-looking statements.
Although forward-looking statements in this Annual Report on Form 10-K reflect the good faith judgment of our Management, such statements can only be based on facts and factors currently known by us. Consequently, forward-looking statements are inherently subject to risks and uncertainties and actual results and outcomes may differ materially from the results and outcomes discussed in or anticipated by the forward-looking statements. Factors that could cause or contribute to such differences in results and outcomes include, without limitation, those specifically addressed under the heading “Risks Factors” below, as well as those discussed elsewhere in this Annual Report on Form 10-K. Readers are urged not to place undue reliance on these forward-looking statements, which speak only as of the date of this Annual Report on Form 10-K. We file reports with the Securities and Exchange Commission (“SEC”). You can read and copy any materials we file with the SEC at the SEC's Public Reference Room at 100 F Street, NE, Washington, DC 20549. You can obtain additional information about the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. In addition, the SEC maintains an Internet site (www.sec.gov) that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC, including us.
We undertake no obligation to revise or update any forward-looking statements in order to reflect any event or circumstance that may arise after the date of this Annual Report on Form 10-K. Readers are urged to carefully review and consider the various disclosures made throughout the entirety of this annual Report, which attempt to advise interested parties of the risks and factors that may affect our business, financial condition, results of operations and prospects.
This Annual Report on Form 10-K includes the accounts of Tonix Pharmaceuticals Holding Corp. (“Tonix”) and its wholly-owned subsidiaries, as follows, collectively referred to as “we”, “us” or the “Company”: Tonix Pharmaceuticals, Inc., a Delaware corporation (“Tonix Sub”) and Krele LLC, a Delaware limited liability company (“Krele”). Tonix Sub is a wholly-owned subsidiary of Tonix and Krele is a wholly-owned subsidiary of Tonix Sub.
Corporate Structure
We were incorporated on November 16, 2007 under the laws of the State of Nevada as Tamandare Explorations Inc. From inception through October 2011, we were involved in the acquisition, exploration and development of natural resource properties in the State of Nevada. On October 7, 2011 (“Closing Date” and the closing of the share exchange transaction, the “Closing”), we executed and consummated a share exchange agreement by and among Tonix Sub and the stockholders of 100% of the equity securities of Tonix Sub, including, the holders of 5,207,500 shares of common stock, 1,500,000 shares of Series A Preferred Stock and 2,275,527 shares of Series B Preferred Stock (the “Tonix Shareholders”), on the one hand, and us and David Moss (“Moss”), our then sole officer and director and majority shareholder, on the other hand (the “Share Exchange Agreement” and the transaction, the “Share Exchange”).
In the Share Exchange, the Tonix Shareholders exchanged their shares of Tonix Sub for newly issued shares of our common stock (“Common Stock”). As a result, upon completion of the Share Exchange, Tonix Sub became our wholly-owned subsidiary.
Upon completion of the Share Exchange, the Tonix Shareholders received an aggregate of 22,666,667 shares of our Common Stock. Moss returned 1,500,000 shares of Common Stock to us, which were retired, and our existing stockholders retained 4,000,000 shares of Common Stock. The 22,666,667 shares issued to the Tonix Shareholders constituted approximately 85% of our 26,666,667 issued and outstanding shares of Common Stock post-Closing.
As a result of the Share Exchange, we acquired 100% of the capital stock of Tonix Sub and consequently, control of the business and operations of Tonix Sub and Krele. From and after the Closing Date of the Share Exchange, our primary operations consist of the business and operations of Tonix Sub and Krele.
On October 11, 2011, we changed our name to Tonix Pharmaceuticals Holding Corp. to reflect our new business.
| 3 |
Corporate Background
In 1996, Seth Lederman, MD, and Donald Landry, MD, PhD, formed L&L Technologies, LLC, (“L&L”), to develop medications for central nervous system (“CNS”) conditions. Dr. Lederman is our Chairman and Chief Executive Officer and Dr. Landry is a Director. L&L was a founder of Janus Pharmaceuticals, Inc., later renamed Vela Pharmaceuticals, Inc., (“Vela”), which developed various therapeutics, including a very low dose, or VLD, version of cyclobenzaprine, under an agreement with L&L. Vela decided to focus its resources on other programs and transferred the rights in VLD cyclobenzaprine and certain other technologies to L&L in March 2006.
Tonix Sub formed in June 2007 as Krele Pharmaceuticals, Inc. by L&L and Krele Pharmaceuticals, LLC (now known as Plumbline LLC) (“Plumbline”). Dr. Lederman is managing partner of Plumbline. Plumbline possessed rights to certain technology for the treatment of alcohol dependence and abuse. In connection with founding Tonix Sub, L&L and Plumbline entered into an intellectual property transfer and assignment agreement with Tonix Sub for the purpose of assigning patents and transferring intellectual property and know-how in exchange for shares of common stock of Tonix Sub. As a result of economic conditions related to the financial crisis of 2007 and 2008, Tonix Sub was not successful in raising money to fund its programs until 2009. As a result, Tonix Sub was unable to advance the development programs and had little activity except for prosecuting and maintaining patents and maintaining contracts.
In 2009, Tonix Sub contracted with the Toronto Psychiatric Research Foundation to analyze the sleep data from the 2001 Phase 2a study of 36 patients with fibromyalgia syndrome, or FM (the “Moldofsky Study”), who were treated with bedtime VLD cyclobenzaprine or placebo. The Moldofsky Study was conducted in Canada by the Toronto Psychiatric Research Foundation, and Tonix Sub obtained the data from this study from L&L. In addition, in 2009, Tonix Sub contracted with Caliper Life Sciences (formerly, NovaScreen Bioscience Corp.) (“Caliper”) to analyze the interactions of cyclobenzaprine with certain receptors. In June 2010, Tonix Sub entered into consulting agreements with L&L and Lederman & Co, LLC (“Lederman & Co”) and also acquired certain rights to develop isometheptene mucate as a treatment for certain types of headaches from Lederman & Co., which we are developing as TNX-201. Dr. Lederman is managing partner of Lederman & Co. Between June 2010 and October 2011, Tonix Sub was active in recruiting new officers and directors and started dosing normal healthy volunteers for the pharmacokinetic trial for TNX-102.
Lederman & Co predominantly provides us with clinical development expertise. L&L predominantly provides us with scientific development expertise. Relative to traditional pharmaceutical development companies, we can be considered a virtual company, since we contract with third-party vendors to provide many functions that are core to traditional pharmaceutical companies. For example, we have contracted with PharmaNet Canada, Inc., or PharmaNet Canada, to develop methods for analyzing cyclobenzaprine in the blood and to conduct a human clinical study to evaluate the performance of our formulation technology. Lederman & Co is responsible for overseeing the scientific and technical aspects of PharmaNet’s contract work product.
In July 2010, Tonix Sub changed its name to Tonix Pharmaceuticals, Inc. In August 2010, Tonix Sub formed Krele.
Business Overview
We are a specialty pharmaceutical company focused on developing new pharmaceutical products for CNS conditions that may be safer and more effective than currently available treatments. We use ongoing advances in science and medicine to search for potential therapeutic solutions among already existing prescription pharmaceutical agents that have been successfully used in patients for other conditions. We create new dose formulations for these agents with the intent to developing products that are optimized for the new therapeutic uses or indications that we target. Our projects are in the development phase, and we currently do not market any products.
The process of taking a new drug formulation from concept through testing to approval for a new indication by the U.S. Food and Drug Administration (“FDA”) is a time-consuming, costly and high-risk process. Once a drug formulation has been tested in laboratories, we need to conduct clinical trials of the product candidate to test its uptake into the blood stream, elimination, effectiveness and safety. Neither laboratory nor animal studies predict the properties of drugs in humans, so designing new formulations can result in a large number of unpredictable outcomes. The first set of clinical trials, which are sometimes referred to as Phase 1 studies, are performed by administering new drug formulations to a limited number of healthy human volunteers and are designed to test the initial concept of the drug formulation and to determine the correct dosage to be tested subsequently on patients affected with the target disorder. The initial Phase 1 studies can take up to a year to complete, however, since we reformulate versions of approved drugs for new uses, we may need to devote less time to Phase I studies since our testing is informed by significant prior human research that we believe allows us to reduce the possible outcomes. The next step in the process is to conduct a larger study in which the new drug formulation is administered to human patients affected with the targeted disorder, which can be referred to as a first pivotal study, a Phase 2b study or a Phase 3 study. The first pivotal study for a condition like FM typically takes a year to complete and then several more months to interpret the data. If the first pivotal study proves the drug is effective and safe, then a second pivotal study is conducted, which can also be referred to as a Phase 3 study. The second pivotal study for a condition like FM would typically take 18 months to complete. After the second pivotal study is completed, and if the results are deemed a success, we would then submit an application to the FDA seeking approval of the new drug product. This application is called a New Drug Application, or NDA. We believe it would take approximately three months to file the FDA application and another 14 months for FDA approval. The drug could be marketed shortly after FDA approval. Therefore, it typically takes more than five years to bring a new formulation of a drug to market for a new indication, and any delays in the process, such as lack of capital necessary to run clinical trials, unexpected results, adverse effects, or difficulty in recruiting willing subjects for trials, would result in additional time before a drug could be available for sale.
| 4 |
Our lead product candidate, TNX-102, is a new optimized dosage form of cyclobenzaprine. TNX-102 is being developed for the management of FM. FM is a CNS condition that is characterized by diffuse musculoskeletal pain, increased pain sensitivity, fatigue and disturbed sleep. Cyclobenzaprine is the active pharmaceutical ingredient of two FDA approved and widely prescribed muscle relaxant products: Flexeril®, an immediate-release form, marketed by the McNeil Specialty Pharmaceuticals division of Johnson & Johnson, and Amrix®, a controlled release form marketed by Cephalon. Generic copies of Flexeril (cyclobenzaprine in the immediate-release form) are available and many patients receive a generic when their physician prescribes Flexeril. Likewise, generic copies of Amrix are also available. According to a study conducted by Frost & Sullivan on our behalf relating to the FM market in the United States (“Frost and Sullivan”), the immediate-release dose form of cyclobenzaprine is widely used off-label to treat FM. We are working to optimize the dose and formulation of TNX-102 to treat FM safely and effectively. We plan to subject TNX-102 to the strict testing required for FDA approval, which we believe will take at least four years and significant clinical studies. We have conducted an initial study of TNX-102 and are currently undertaking a comparative pharmacokinetic and bioavailability study, of which the clinical phase was completed by the end of 2011, and the analysis of the subjects’ blood samples will be completed in the first half of 2012. If TNX-102 is ultimately approved by the FDA for the management of FM, we believe it will be adopted by physicians and reimbursed by managed care companies.
Our other leading product candidate, TNX-105, which we are also developing, is a new dose form of cyclobenzaprine to treat symptoms of post-traumatic stress disorder, or PTSD. PTSD is a psychiatric disorder that begins in the aftermath of traumatic experiences. Sleep disturbances, including nightmares and insomnia, are core features of PTSD and are included in two of the three main symptom clusters. Patients with PTSD may have any single or combination of symptoms that include re-experiencing, emotional numbing and avoidance, and hyperarousal reactions that persist for more than one month after the traumatic event. PTSD shares several features with FM, and some patients are believed to suffer from both PTSD and FM.
Cyclobenzaprine is the active pharmaceutical ingredient in each of our lead product candidates. We are utilizing drug delivery technology to produce new formulations. In addition to cyclobenzaprine, each formulation of TNX-102 and TNX-105 will contain inactive ingredients, called excipients that are well-characterized and have been FDA approved previously in other products. As a result, we anticipate seeking FDA marketing approval of our lead product candidates, TNX-102 and TNX-105, through the NDA process under Section 505(b)(2) of the U.S. Federal Food, Drug and Cosmetic Act, or the FFDCA, which we also refer to as Section 505(b)(2). This process permits the FDA to make some safety and effectiveness determinations through review of materials in the public domain or in already approved NDAs of products containing cyclobenzaprine. This approach would spare us some of the burden of generating all of this data for ourselves and may allow our lead product candidates to progress through a shorter development pathway than is typical for pharmaceutical products based on novel active ingredients. We have not filed an NDA for either of our lead product candidates.
We also have a pipeline of several other product candidates that we are constantly evaluating. For example, we are developing TNX-201, which is a treatment for certain types of headaches and TNX-301, which is a potential treatment for alcohol dependence and addiction. For commercial reasons, we normally do not disclose the identities of the active ingredients or targeted indications of products in our pipeline until a U.S. patent has been allowed. Consistent with our mission, these product candidates are, or likely will be, reformulations of active ingredients that have been used by patients in other FDA-approved products. We anticipate that some of our other pipeline products will be submitted to the FDA for approval under Section 505(b)(2). In other cases, we expect that the products will be formulated to match earlier predicate products closely enough to rely, in part, on their regulatory review and status. There may be instances where the predicate product is a medicine that was reviewed for safety and effectiveness by the National Academy of Sciences under the Drug Evaluation and Safety Initiative, or DESI, and would be considered by the FDA to be an “unapproved product.” For DESI products, it is our intent also to develop NDA versions by modernizing the chemistry, manufacturing and controls and to perform new clinical studies to support an NDA filing under Section 505(b)(2).
Because of our size and being in the development stage, we do not currently devote a significant amount of time or resources towards our other pipeline candidates. We anticipate that sometime in 2012 we will begin developing formulations for TNX-201 and possibly TNX-301, but do not expect to start clinical trials until 2013 at the earliest.
| 5 |
Krele’s mission is to commercialize products that are generic versions of predicate NDA products or existing marketed products that it may acquire from other pharmaceutical companies. We expect that Tonix Sub’s relationship to Krele will be similar to that of several other pharmaceutical companies and their subsidiaries that market generic versions of the parent’s branded products at different periods in their product life-cycle. We anticipate that when one of our branded products loses patent protection, Krele may market generic versions of it. In such instances, Krele’s product would be an “authorized generic” and would rely on our NDA. Krele may also develop or acquire generic products approved under Abbreviated New Drug Applications (“ANDAs”). For ANDAs, the predicate product is a medicine approved by the U.S. Food and Drug Administration (the “FDA”) under an NDA. Tonix Sub may market branded versions of such products that rely on Krele’s ANDAs which would be referred to as branded generics. We do not currently market any products and have only begun the process of obtaining state licenses, which are legally required before a company can manufacture, distribute and market prescription medications. Krele has been issued a state license in New York.
Our Strategy
Our objective is to develop and commercialize our product candidates to treat CNS conditions, including FM and PTSD. The principal components of our strategy to achieve this objective are to:
| · | pursue development and regulatory approval pathways by reformulating versions of approved drugs for new uses and by using the Section 505(b)(2) pathway for FDA approval; | |
| · | adopt a two-pronged patent strategy by seeking methods of use patents for the active ingredients in our products and by seeking protection for the formulation technology employed in our products; | |
| · | provide clear value propositions to third-party payers, such as managed care companies or government programs like Medicare, to merit reimbursement for our product candidates; and | |
| · | enter into collaborations with other pharmaceutical companies with respect to, among others, our FM and PTSD product candidates and other products that will benefit from development or marketing resources beyond those in our Company. |
Pursue development and regulatory approval pathways. We believe our lead product candidates may be approvable under pathways that are potentially shorter than those typically available for drug products based on novel active ingredients. By focusing on developing new formulations of approved drugs for new uses, we believe that we will be able to use the Section 505(b)(2) pathway for FDA approval. This pathway can reduce the time and expense required for our development programs by allowing our use of previously-generated safety and efficacy information regarding the active pharmaceutical ingredients in our lead product candidates to support the filing and approval of our NDA application. Doing so may help reduce the size and scope of our preclinical and clinical trials. The FDA has strict requirements on the marketing of drugs, and FDA approves each drug for specific uses which are called the label indications. The use of a drug product for the treatment of a condition other than one of its approved label indications is called off-label use. The development of an existing FDA-approved drug for the treatment of a condition other than one of its approved label indications is considered a “new use”. For companies involved in the ethical development and marketing of prescription drugs in the US, FDA approval of a new use or new label indication is the only legal basis of marketing claims. Off-label use is not recognized by the FDA or FDA-regulated companies as a new use.
Adopt a two-pronged patent strategy. We are pursuing a two-pronged patent strategy by seeking intellectual property protection for our methods of use for certain known active pharmaceutical ingredients and by seeking patents to protect the formulation technologies we employ. With respect to the methods of use patents, we believe the therapeutic uses we target are new uses for these active ingredients and we have been issued patents directed to certain aspects of our new uses. We are seeking additional patents to cover other new uses. For example, the invention of bedtime VLD cyclobenzaprine as a treatment for FM was novel and unexpected when our patents were filed in 2000. With respect to formulation patents, we believe our products will be protected by patents that describe inventions of technology for making new formulations and possibly also by patents that describe the invention of products that achieve novel and useful blood levels at certain times after administration.
Provide clear value propositions to third-party payors to merit reimbursement for our product candidates. We are designing our clinical development programs to demonstrate compelling competitive advantages to patients and prescribers and also to demonstrate value propositions to third-party payors. We believe TNX-102 might help in the management of FM by reducing pain and other symptoms, such as fatigue. In addition, we believe that bedtime treatment with TNX-102 will have fewer day time side-effects than off-label bedtime treatment with immediate release cyclobenzaprine. For FM, we believe an FDA-approved product would capture some of the off-label use of generic cyclobenzaprine. Because FDA approvals are based on objective data, we believe that third-party payors will provide reimbursement for an FDA approved product, even at a premium price relative to other drugs that are used off-label, such as immediate-release cyclobenzaprine, tizanidine, baclofen, carisoprodol or metaxalone. For example, third-party payors reimburse for using FDA approved Lyrica® and Cymbalta® for fibromyalgia over off-label generic versions of Neurontin® (gabapentin) and generic anti-depressants, respectively.
| 6 |
Enter into collaborations to maximize the value of our technology. We believe certain of our drug development candidates, including TNX-102 and TNX-105, can be developed and marketed more effectively by companies that already have significant drug development and marketing capabilities. We will seek to enter into collaborations with pharmaceutical or biotechnology companies for the commercialization of these product candidates at the times we believe most effective.
Our Lead Product Candidates
Our lead product candidates are TNX-102, for the treatment of FM and TNX-105 for the treatment of PTSD. Both of these consist of cyclobenzaprine in a mixture of inactive ingredients that are called “excipients”, which we believe will improve the absorption rate of cyclobenzaprine in ways that will optimize the product for bedtime treatment.
Cyclobenzaprine
Cyclobenzaprine was first synthesized in 1961 by Merck, and the 10 mg Flexeril immediate-release dose form was FDA approved in 1977 for the relief of muscle spasm associated with acute, painful musculoskeletal conditions as an adjunct to rest and physical therapy.
Although a number of clinical studies have addressed the potential use and benefit of cyclobenzaprine in treating symptoms of FM, to our knowledge these studies have not motivated a sponsor to pursue FDA approval.
Based on cyclobenzaprine’s safety and efficacy for treating muscle spasm, in the 1990s, Merck conducted studies to support an application to market a 5 mg Flexeril tablet (low dose) for the over-the-counter, or OTC, market, where patients can purchase medicine without a physician’s prescription. Although Merck’s studies re-affirmed the safety and demonstrated efficacy of 5 mg Flexeril in several large trials, the OTC division of the FDA rejected the application for use without a prescription, apparently, we believe, because muscle spasm was deemed a condition that required a physician to diagnose and supervise treatment.
Merck divested the Flexeril franchise to Alza Pharmaceuticals, or Alza. Alza subsequently was acquired by Johnson and Johnson and Flexeril is part of their McNeil Specialty Pharmaceuticals division. Based largely on the Merck studies, McNeil won approval of Flexeril 5 mg tablets as a prescription medicine to treat muscle spasm. McNeil promoted Flexeril 5 mg tablets for the three year period of market exclusivity based on The Drug Price Competition and Patent Term Restoration Act of 1984, generally referred to as the Hatch-Waxman Act. Following this exclusivity period, several generics entered the market and took market share from Flexeril. McNeil continues to manufacture Flexeril, but we believe McNeil no longer actively promotes it.
Despite the approved uses of cyclobenzaprine in treating muscle spasm, we believe current marketed formulations of cyclobenzaprine are limited for treating FM by unpredictable absorption. As described in the Flexeril package insert, the amount of cyclobenzaprine absorbed into the bloodstream varies between 33-55% of the dose ingested. The variability in absorption may be due to several factors, including effects of the stomach pH (acidity or base) on the dissolution of the tablets, as well as the context of either an empty stomach or a recent meal. Food in the stomach and small intestine from a recent meal contributes to variability in absorbing other drugs. The uncertainties in absorption rates make it challenging for a physician contemplating a bedtime treatment for FM to ensure the intended therapeutic effect is achieved without risking side effects like next-day drowsiness, which could result if the patient has too much cyclobenzaprine remaining in the bloodstream the next day.
If a product could deliver a predictable absorption rate of cyclobenzaprine, it would mean patients would be less likely to receive too little drug to receive a therapeutic effect. Conversely, patients would be less likely to be over-dosed, which might lead to potential side effects, including next-day drowsiness. An optimal VLD cyclobenzaprine product could have faster absorption, faster clearance and more predictable effects than the immediate release tablet format. We are testing a number of technologies to optimize the properties of TNX-102 for FM and TNX-105 for PTSD. One of the technologies we are testing is a novel gelatin capsule (gelcap) that employs a proprietary mixture of lipids with cyclobenzaprine. The proprietary lipid mixture is designed to increase the rate and efficiency of absorption of cyclobenzaprine from the gastrointestinal tract into the bloodstream. This formulation is expected to result in increased dosage precision. However, the science of formulating drugs is not sufficiently advanced to predict the performance of the new gelcaps or other formulation technologies in humans. We will only learn if our design has advantageous properties when we conclude testing of TNX-102 in human subjects.
TNX-102 in Fibromyalgia Syndrome
TNX-102, our most advanced product candidate, is a bedtime pill containing VLD cyclobenzaprine (2.4 mg). The development of TNX-102 is supported by the results of the Moldofsky Study of VLD cyclobenzaprine in FM patients. A version of TNX-102 has been manufactured in small quantities for use in human clinical trials. Based on our formulation of TNX-102, we are testing whether it will provide more predictable effects and decreased risk of next-day drowsiness than commercially available immediate-release cyclobenzaprine tablets. We are testing a variety of technologies for faster and more efficient absorption relative to currently marketed cyclobenzaprine products.
| 7 |
FM is diagnosed by groups of symptoms that have been defined by committees of the American College of Rheumatology, or ACR, and a committee of experts from the organization Outcome Measures in Rheumatology. In 2007, Pfizer’s Lyrica (pregabalin) became the first medicine approved by the FDA for the management of FM. In 2008, Eli Lilly’s Cymbalta (duloxetine) became the second medicine approved by the FDA for the management of FM. In 2009, Savella® (milnacipran) was the third medicine approved by the FDA for the management of FM. Savella is marketed by Forest Laboratories.
Product Development Path
Phase 2a Pilot Data in FM Patients
Our motivation to focus our efforts on developing TNX-102 for FM stems from the results of a clinical study on 36 patients in 2001, the related rights to which we acquired from L&L. Specifically, this study was a randomized, double-blind, placebo-controlled, dose-escalating eight week trial conducted at two study centers. The study subjects met ACR criteria for FM.
Patients received VLD cyclobenzaprine immediate-release 1 mg capsules or corresponding placebo capsules to ingest after dinner and before bedtime. Initially, patients took one capsule each evening, but over the course of the study, they were allowed to increase the number of tablets taken in increments of one capsule per week. The maximum number of capsules allowed was four per evening, which would be a total dose of 4 mg immediate-release cyclobenzaprine.
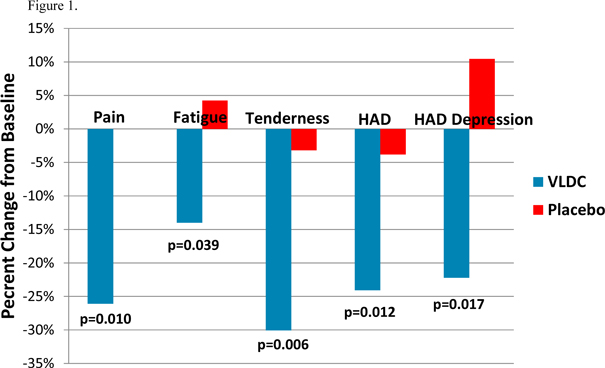
Patients treated with VLD cyclobenzaprine demonstrated significant improvements in pain, fatigue and tenderness at week 8 relative to baseline whereas, placebo-treated patients did not improve (Figure 1). Although this study excluded patients who met formal criteria for major depressive disorder or any anxiety disorder, there is a high degree of co-existing symptoms of depression and anxiety associated with FM. VLD cyclobenzaprine treatment resulted in significant reductions in total Hospital Anxiety and Depression Scale, or HAD, which measures symptoms of anxiety and depression, and the HAD depression subscale which measures depressive symptoms (Figure 1).
This study showed treatment with VLD cyclobenzaprine:
| · | provided benefit in core symptoms of FM, including pain and fatigue; |
| · | improved mood, by demonstrating a significant decrease in HAD scores; and |
| · | was well tolerated, with no serious adverse events, or SAEs, or discontinuations due to adverse events, or AEs. |
| 8 |
Pharmacokinetic Study
We have conducted a human clinical study being conducted by a contract research organization, or CRO, under an US Investigational New Drug Application, or IND, and a Canadian Clinical Trial Application. We received FDA and Health Canada clearance for this study, which was conducted in Canada. This study will determine the blood levels of cyclobenzaprine in approximately 30 healthy adult volunteers after they ingest either TNX-102, a candidate gelcap formulation containing low-dose cyclobenzaprine or a currently marketed, immediate-release cyclobenzaprine product. Studies that measure the blood levels of drugs over time are called “pharmacokinetic studies”. The TNX-102 formulation is being tested in subjects who are either fasting or recently fed. This study seeks to measure the circulating blood levels of cyclobenzaprine after oral administration of the TNX-102 candidate formulation in a fed or fasting state and determine how they compare to the blood levels resulting from oral administration of the currently marketed product in a fasting state. Each subject receives each of the trial doses and conditions in a random order, in what is called a crossover study design. The crossover design allows the assessment of the variability of drug blood levels over time in the same people during each phase. We selected PharmaNet Canada to conduct this pharmacokinetic study. The clinical portion of this study was completed by the end of 2011, and we expect to finish analyzing the specimens and interpret the data in the first half of 2012. The study is expected to cost approximately $1 million, which includes the cost of manufacturing TNX-102 and placebo.
Prospective Phase 2b Study
If our pharmacokinetic study is successful, we expect to advance the clinical development of TNX-102 for the management of FM by conducting a larger Phase 2b placebo-controlled study. Utilizing our proprietary formulation, we will seek to replicate and expand upon the efficacy and safety findings of our Moldofsky Study by administering the commercial form of TNX-102 or placebo to approximately 300 FM patients for twelve weeks. We expect that our proposed Phase 2b will be one of the two clinical efficacy trials required for FDA approval.
We expect the outcome measures for efficacy in this study will be similar to those utilized by drug products currently approved for use in FM. Specific efficacy outcome measures will include the Brief Pain Inventory, the Patient Global Impression of Change (PGIC) and the Fibromyalgia Impact Questionnaire (FIQ). Additional outcome measures for this trial will be carefully planned to further our exploration of treatment effects in important areas such as sleep, fatigue, mood, sexual function and quality of life. We will seek FDA concurrence on the study design and expect to engage a CRO to conduct this study on our behalf. We expect the study will enroll a first patient in the third quarter of 2012 and will be completed in the third quarter of 2013. We anticipate this study will cost approximately $15 million, which includes the cost of manufacturing TNX-102 and placebo.
Prospective Multi-dose Pharmacokinetic Study
Since cyclobenzaprine will be used chronically in TNX-102, we will study TNX-102 in comparison to immediate-release cyclobenzaprine in multiple day dosing (once daily). Subjects will ingest TNX-102 or immediate-release cyclobenzaprine for four or more days. Peak and trough blood levels of cyclobenzaprine will be measured. The results of this study will provide information regarding blood levels of cyclobenzaprine when taken in a multiple day regimen.
Prospective Study Comparing Side-effects of TNX-102 with Immediate-Release Cyclobenzaprine
We plan to conduct a small study designed to compare the bedtime use of TNX-102 and immediate-release cyclobenzaprine on next morning drowsiness. The goal of this study is to determine the potential benefit of TNX-102 compared with immediate-release cyclobenzaprine on next morning drowsiness.
Prospective Phase 3 Study
If our Phase 2b study is successful, then we expect to conduct a Phase 3 study in support of product registration. At this time, we plan to conduct one large scale, randomized, double-blind, placebo-controlled Phase 3 study in which patients with FM will receive TNX-102 or placebo for six months. It is likely that the outcome measures for efficacy in this study will be similar to those used in the Phase 2b study. Other outcome measures will be carefully considered to best support desired label claims and optimal marketing message for product differentiation. We expect that at least 300 FM patients will be enrolled in this trial.
Safety Exposure Study
To study the safety of our product in chronic use, we expect to conduct an open label study in which approximately 300 FM subjects would receive TNX-102 for up to one year. Together with our other studies, we believe this safety exposure study will support the FDA and international regulatory requirements to provide data for at least 300 subjects treated with TNX-102 for six months and at least 100 subjects treated for 1 year.
| 9 |
Regulatory Strategy
The approvals of Lyrica, Cymbalta and Savella establish a regulatory approval standard for management of FM. However, given the heterogeneity of patients with this disease, it may not prove to be the only pathway or approval requirement. Prior to meeting with the FDA for an End-of-Phase 2 (EOP2) meeting, we plan to strategically assess the regulatory environment and further evaluate our Phase 2 results in order to determine the optimal design of phase 3 clinical program. The phase 3 study design will be discussed with the FDA at the EOP2 meeting to receive regulatory acceptance for a differentiated product for the management of FM.
We hope to register TNX-102 with the FDA through the provisions of Section 505(b)(2). This regulatory pathway may help to accelerate product development and reduce overall business risk. The 505(b)(2)-based product development plan for TNX-102 is designed to leverage the safety data that has been generated by other manufacturers for cyclobenzaprine-containing products and accepted by the FDA in support of their product registration. TNX-102 contains significantly less active cyclobenzaprine than other marketed products. We believe that the safety data package from these products will provide adequate safety margin to support TNX-102 development.
On August 11, 2011, we had a pre-IND meeting with the Division of Anesthesia, Analgesia and Addiction Products within the Center for Drug Evaluation and Research at the FDA to discuss the IND and NDA requirement of TNX-102 for the management of FM. Based on the meeting outcome, we successfully filed an IND application on October 10, 2011, which received FDA clearance on our IND study on November 10, 2011. The planned IND study is conducted in Canada under a Canadian Clinical Trial Application filed October 7, 2011, which received “No Objection Letter” on November 7, 2011. We will continue working with the FDA to seek guidance and agreement on the TNX-102 development program, specifically the necessary data to support the 505(b)(2) NDA regulatory pathway. As FDA indicated at the pre-IND meeting, the clinical trials in our development plan, if successful, will provide efficacy and safety data sufficient to support an NDA filing.
If NDA approval is granted for TNX-102, in addition to the 3-year marketing exclusivity granted, TNX-102 is expected to be covered under patents that extend through at least 2021, during which time it should not be subject to generic substitution. We plan to continue to support the TNX-102 program with new patent applications as we obtain data from the clinical evaluation of our new formulation in healthy human subjects and FM patients.
TNX-105 in Post-traumatic Stress Disorder
TNX-105, our second most advanced product candidate, is another pill formulation of cyclobenzaprine to be taken at bedtime for PTSD, a psychiatric disorder that begins in the aftermath of traumatic experiences. We have not yet conducted any clinical trials on PTSD patients.
Parallels Between FM and PTSD
A number of parallels have been noted between FM and PTSD. In addition, symptom overlaps may exist between patients diagnosed with FM or PTSD. In a survey of males with PTSD or major depression (Amital, Fostick et al, Posttraumatic stress disorder, tenderness, and fibromyalgia syndrome: are they different entities? J. Psychosom Res 2006. 61(5):663-9.2006), 49% of PTSD patients met the ACR criteria for FM compared to 5% of major depression patients. Conversely, in a different survey of FM patients (Cohen, Neumann et al., Prevalence of post-traumatic stress disorder in fibromyalgia patients: overlapping syndromes or post-traumatic fibromyalgia syndrome? Semin Arthritis Rheum 2002. 32(1):38-50), 57% of the sample had symptoms associated with PTSD.
Emerging Market Opportunity
The selective serotonin reuptake inhibitors Paxil® (paroxetine) and Zoloft® (sertraline) are FDA approved for PTSD, but are not satisfactory treatments for many patients. Other drugs that show promise for the treatment of PTSD, but are not FDA approved, include antidepressants such as nefazodone, mirtazapine and trazodone; the antihistamine cyproheptadine; certain atypical antipsychotics such as olanzapine and risperidone; and an adrenergic alpha-1 receptor blocker, prazosin. Prazosin may decrease nightmares and insomnia and has been associated with improvements in daytime PTSD symptoms, depression, and quality of life.
Our rationale for studying the effects of cyclobenzaprine in PTSD derives from the following:
| · | our clinical studies that very low dose cyclobenzaprine improves FM symptoms, a disorder having significant overlap with PTSD; and |
| 10 |
| · | in studies conducted by Caliper, cyclobenzaprine interacts with a receptor on brain cells called the serotonin type 2a receptor. Based on numerous peer-reviewed scientific publications, we have identified a number of compounds that bind this receptor that have been shown to have effects in treating PTSD. Therefore, it is our belief that cyclobenzaprine, because it binds to the serotonin type 2a receptor, will have a therapeutic effect in treating PTSD like other compounds that bind to it. |
In 2009, we engaged Caliper to learn which receptors in the brain bind cyclobenzaprine. Caliper measures the interactions of receptors with active pharmaceutical ingredients and has built a proprietary database. Arthur Weissman, PhD is Vice President and Chief Scientific Officer at Caliper and supervised the receptor study. Dr. Weissman holds a M.S. degree in Physiology, a Ph.D. degree in Neuroscience, has over 25 years of scientific research and has authored (or co-authored) over 20 peer-reviewed scientific publications. The receptor studies were conducted at Caliper’s facilities. Caliper is constantly conducting receptor studies to expand and refine its database, so the date of each individual receptor-drug analysis is different. Caliper provided us proprietary data from their database, which showed cyclobenzaprine binds to the serotonin type 2a receptor.
Product Development Path
Prospective Phase 2a and 2b Studies
We anticipate that the dose for treatment of PTSD symptoms may be higher than that of TNX-102 for FM. We plan to utilize the data obtained from the pharmacokinetic study of TNX-102 to design a Phase 2a study for TNX-105. We expect that this study will employ the same formulation technology used for FM, but will be dosed with multiple pills to explore a dose range for efficacy and tolerability in PTSD. The estimated treatment period will be six to eight weeks in duration.
As part of our contemplated Phase 2a study, we plan to assess the appropriateness of a number of clinical outcomes for use as primary and secondary measures. The PTSD clinical study measures used for further development work must provide adequate specificity and sensitivity to measure the potential effects of cyclobenzaprine. In our Phase 2a study, we anticipate that we will study TNX-105 in less than 50 subjects with combat-related and/or civilian PTSD. We expect to engage a CRO to conduct this study on our behalf.
After exploring the clinical utility and dose range in a Phase 2a study, we intend to advance the clinical development of TNX-105 for the treatment of PTSD by conducting a larger randomized, double-blind, placebo-controlled study in Phase 2b. The treatment period is estimated to be eight to twelve weeks in duration. We will seek to replicate and expand upon the efficacy and safety findings of the Phase 2a study in a larger population of PTSD patients. In our Phase 2b study, we anticipate that we will study the drug in 100 to 150 subjects with combat-related and civilian PTSD. We expect to engage a CRO to conduct this study on our behalf.
Prospective Phase 3 Study
If our Phase 2b study is successful, we expect to conduct a Phase 3 program in support of an NDA. At this time, our general plan includes two large scale, randomized, double-blind, placebo-controlled Phase 3 studies, and one open-label extension study. We anticipate that the treatment duration for the two large studies will be approximately 12-16 weeks in length. The numbers of patients to be evaluated is unknown at this time. We plan to confer with the FDA concerning the suggested sample sizes in an End-of-Phase 2 program review meeting. Once completing their participation in one of the two large scale studies, we expect our subjects will have the choice of enrolling in an available open-label study whereby we can assess the longer-term benefits of TNX-105 therapy in PTSD.
Regulatory Strategy
The approvals by the FDA of Paxil (paroxetine) and Zoloft (sertraline) for treating PTSD establish a regulatory approval pathway for symptom reduction in PTSD. We plan to strategically assess the regulatory environment and further evaluate our Phase 2 results to determine the design of Phase 3 clinical studies. We believe these studies will result in a differentiated product for the treatment of PTSD. We hope to register TNX-105 with the FDA through the provisions of Section 505(b)(2).
We anticipate meeting with the Center for Drug Evaluation and Research at the FDA to discuss TNX-105 at the appropriate time in the future and would review the basis of our Section 505(b)(2) clinical development plan and discuss any other clinical and nonclinical trials necessary to support an NDA filing. We believe that the clinical trials in our development plan, if successful, will satisfy the requirements for sufficient evidence of clinical efficacy and safety to support an NDA.
TNX-105 is expected to be covered under patents that have been submitted to the USPTO. The USPTO has not yet allowed or granted any claims protecting the use of TNX-105.
| 11 |
Drug Delivery Technology
We are investigating different technologies to improve the absorption of cyclobenzaprine. For example, we identified and obtained an exclusive worldwide option on technology from Lipocine, Inc. (“Lipocine”) that employs mixtures of different types of lipids. Under our agreement, Lipocine studied a number of combinations of lipids for their ability to form micelles that solubilize the free base of cyclobenzaprine and which might serve as inactive ingredients in a gelatin capsule formulation. We selected a candidate formulation based on properties that included the dispersion of the active ingredient in simulated gastric or small-intestinal fluids and the stability of the formulation over time prior to testing. Lipocine was also engaged to manufacture gelatin capsules of TNX-102 for use in our pharmacokinetic trial. In the option agreement, we agreed to make certain sublicense, royalty and milestone payments, which are subject to a request for confidential treatment. Pursuant to the agreement, we have an option to license Lipocine’s US patent 6,294,192 “Triglyceride-free compositions and methods for improved delivery of hydrophobic therapeutic agents” and US Patent 6,451,339 “Compositions and methods for improved delivery of hydrophobic agents”. These patents expire on September 24, 2021 and September 16, 2022, respectively. If we exercise the option to license these patents, we will be obligated to pay Lipocine low single-digit percentage royalties based on net sales or mid-teen sublicense fees based on the consideration that we receive from a licensee.
Both of our cyclobenzaprine-based product candidates consist of cyclobenzaprine in pills that also contain proprietary ingredients, that are inactive but help the small intestine absorb cyclobenzaprine. TNX-102 and TNX-105 are formulations of cyclobenzaprine and mixtures of lipids that are intended as bedtime treatments for FM and PTSD, respectively.
We have concluded a study of the stability and dissolution of several candidate formulations in simulated gastric and small-intestinal fluids. The study was conducted in 2007 at Lipocine’s facilities. The first element of the study was to screen lipid ingredients for use in a gelcap. In this study, various lipid ingredients were mixed with cyclobenzaprine to determine solubility and suitability for formulating cyclobenzaprine in gelatin capsules, or gelcaps. Based on the results of the screening, four formulations of cyclobenzaprine hydrochloride were prepared and analyzed for how efficiently they released or dispersed cyclobenzaprine into solutions of simulated gastric and small-intestinal fluid. Each of the four formulations resulted in about 95% or more of cyclobenzaprine in solution. Three of four formulations rapidly (at 30 minutes) released more than 90% of cyclobenzaprine into an acidic solution that simulates gastric conditions. The second element of the study evaluated physical stability of the formulations. The four candidate formulations were filled into capsules and subjected to stability conditions at high temperature and temperature cycling. None of the four formulations showed signs of phase separation or crystallization of cyclobenzaprine. All four formulations were stable and none showed signs of reduction in cyclobenzaprine potency compared to the initial time. From these data, we selected two potential formulations for further study based on solubility level and speed of dissolution in acid. Results from this study showed that certain proprietary lipid mixtures interact with cyclobenzaprine to help solubilize it in simulated gastric and small-intestinal fluids. Based on the study, we have selected a candidate formulation for cyclobenzaprine to be dosed at bedtime.
We expect TNX-102 and TNX-105 will employ the same formulation, but TNX-105 will contain a higher dose of cyclobenzaprine. We believe one or more of our new formulations will result in the more efficient and more predictable cyclobenzaprine absorption than immediate-release cyclobenzaprine tablets that are commercially available for daytime use to treat muscle spasm. Since we expect our formulations will be more efficiently absorbed, we believe lower doses of cyclobenzaprine in our proprietary formulations with lipids will provide a similar therapeutic benefit to higher doses of immediate-release cyclobenzaprine.
Market Dynamics
We believe the U.S. market for products that treat CNS conditions has several characteristics that make it an attractive market for pharmaceuticals, including that the customer base is driven by physicians who are involved in long-term care of patients with chronic disorders. Patients with CNS disorders sometimes carry disease burdens that require long-term treatment.
We believe the market for FDA-approved FM treatments is underserved and that there is a constant need for new treatment options, since many prescription drugs provide relief only to some of the affected patients or provide relief only for limited periods of time.
Until 2007, there were no FDA-approved drugs to treat FM. A number of effective medicines have been identified by physicians who observe improvements in a patient’s condition as an unintended consequence of prescribing a particular medicine for another purpose. These anecdotal observations are sometimes substantiated by exposing additional patients in progressively more systematic studies. As information about a potential benefit is reported in scientific literature, or shared among physicians, an increasing number of physicians may prescribe such medicines to their patients. This practice, which is not sanctioned by the FDA, is referred to as “off-label” prescribing or use. Off-label prescription practices in the U.S. are acceptable under a long-standing principle that grants physicians the ability to use their professional judgment beyond the FDA recommended uses.
| 12 |
Before 2007, a variety of drugs, often in combination, were utilized off-label to treat symptoms associated with FM. The following three classes of drugs were prescribed as the primary treatments for FM: (1) pain killers, also referred to as analgesics, (2) antidepressants and (3) muscle relaxants.
In 2007, Lyrica (pregabalin) became the first medicine approved by the FDA for the management of FM. Lyrica previously had been approved and marketed to treat pain in other conditions. FM shares a number of symptoms with depression, and a number of FM patients are believed to experience depression as a co-existing condition. In 2008, Cymbalta (duloxetine) became the second medicine approved by the FDA for the management of FM. Cymbalta previously had been approved and marketed to treat depression. Savella (milnacipran) was the third medicine approved by the FDA for the management of FM. Savella’s active ingredient, milnacipran, is approved in Europe to treat depression.
Since Lyrica and Cymbalta also are marketed for other conditions beyond FM, the sales of these products related specifically to FM can only be estimated. According to Frost & Sullivan, the overall gross sales for FM prescription drugs in 2010 was believed to be about $1.2 billion, which has grown since 2007 at a compounded annual growth rate of 18.4%. This significant increase is a result of more FM patients switching to branded FM prescription drugs that sell for a higher cost than the generic FM prescription drugs previously used. For example, in 2010, Lyrica prescriptions are estimated to have accounted for 248 million doses for FM and to have generated $478 million in sales, while Cymbalta prescriptions are estimated to have accounted for 93 million doses for FM and to have generated $342 million in sales. Launched in January 2009, Savella, which is only approved for the treatment of FM, prescriptions accounted for approximately 43 million doses and generated approximately $68 million in sales in 2010.
Use of the FDA approved medications for FM is growing while the use of off-label treatments is declining. Overall, in terms of the number of doses of FM prescription drugs prescribed, Frost & Sullivan expects the FM market to grow at only a 1.2% compounded annual growth rate from 2007 to 2010. These market dynamics are consistent with the interpretation that Lyrica’s growth came at the expense of off-label pain killers and Cymbalta’s and Savella’s growth came at the expense of off-label anti-depressants.
According to Frost and Sullivan, FM is an emerging market and sales are anticipated to continue growing in future years. Despite the availability of FDA approved products, we believe the current treatment options for FM continue to leave many patients dissatisfied.
The FM market for muscle relaxants lacks an FDA-approved product and continues to be satisfied by off-label medicines such as cyclobenzaprine, tizanidine, baclofen, carisoprodol and metaxalone. These muscle relaxants have generic and branded versions. According to Frost & Sullivan, 48 million doses of the Flexeril brand and its associated immediate-release cyclobenzaprine generic products were prescribed off-label for FM in 2010 and accounted for approximately 35% of the muscle spasm pills prescribed for FM. However, the off-label cyclobenzaprine sales for FM in terms of dollars amount to only approximately $10 million, due to the low price of generic cyclobenzaprine.
Challenges in the Market for CNS Therapies
Developers of pharmaceutical treatments for syndromes and disorders that affect the CNS face special challenges. In many cases, the causes and exacerbating factors of CNS conditions remain unknown. Frequently, key symptoms are known only by patient reports and cannot be objectively validated or measured. Symptoms like pain, fatigue, disturbed sleep or altered mood are characteristics of more than one condition. Often, physicians may not agree that a particular patient is affected by one or another condition or by more than one co-existing conditions.
CNS conditions are typically defined by committees of expert professionals who set criteria based on the presence of several symptoms or groups of symptoms. Sometimes groups of subjective symptoms are insufficient to describe CNS disorders and further refinement of diagnostic categories can be achieved by patient demographics, such as gender, age or concurrent medical processes, such as menopause or adolescence. Many CNS conditions, including syndromes and disorders, have not yet been characterized by laboratory tests, such as blood tests or x-ray imaging. However, laboratory tests are often important to exclude other conditions, such as inflammatory or infectious processes. Consequently, a CNS condition is sometimes called a diagnosis of exclusion because inflammation and infection should typically be ruled out by laboratory tests before applying the criteria of groups of symptoms to diagnose it.
Once a CNS condition is diagnosed, physicians may select from among treatment options based on a patient’s symptoms and history. Some medications improve or relieve only one or another symptom in a condition. Consequently, physicians may prescribe several different medications concurrently to treat individual symptoms or groups of symptoms. A desirable quality for CNS medications is the ability to relieve more than one symptom of a CNS condition. Another desirable quality for CNS medications is safety, particularly if a medicine is safe enough to be used with other medicines concurrently or at different times of the day.
| 13 |
Opportunity for New Treatments of FM
We believe the market for the treatment of FM is underserved which we believe fuels a need for new therapeutic options. Due to the market acceptance of FM treatments (such as Lyrica, Cymbalta and Savella), we believe there will be a growing interest in alternative drug treatment options.
We believe that if TNX-102 won FDA approval, it would be an appealing option because it has an entirely different mechanism of action from the currently approved products and we expect TNX-102 will be recommended for use before bedtime. Lyrica is recommended for twice or three-times daily dosing. Cymbalta was found effective at once-daily dosing and is generally restricted to daytime use and not recommended for bedtime use. Cymbalta and Savella act on the CNS in ways that are believed to interfere with sleep.
Competition
Our industry is highly competitive and subject to rapid and significant technological change. Our potential competitors include large pharmaceutical and biotechnology companies, specialty pharmaceutical and generic drug companies, academic institutions, government agencies and research institutions. We believe that key competitive factors that will affect the development and commercial success of our product candidates are efficacy, safety, tolerability, reliability, price and reimbursement level. Many of our potential competitors, including many of the organizations named below, have substantially greater financial, technical and human resources than we do and significantly greater experience in the discovery and development of product candidates, obtaining FDA and other regulatory approvals of products and the commercialization of those products. Accordingly, our competitors may be more successful than we may be in obtaining FDA approval for drugs and achieving widespread market acceptance. Our competitors’ drugs may be more effective, or more effectively marketed and sold, than any drug we may commercialize and may render our product candidates obsolete or non-competitive before we can recover the expenses of developing and commercializing any of our product candidates. We anticipate that we will face intense and increasing competition as new drugs enter the market and advanced technologies become available. Further, the development of new treatment methods for the conditions we are targeting could render our drugs non-competitive or obsolete.
The markets for medicines to treat FM, PTSD and other CNS conditions are well developed and populated with established drugs marketed by large and small pharmaceutical, biotechnology and generic drug companies. Pfizer (Lyrica), Eli Lilly (Cymbalta) and Forest Laboratories/Cyprus Biosciences (Savella) market FDA approved drugs for FM. Pfizer (Zoloft) and GlaxoSmithKline (Paxil) market FDA approved drugs for PTSD.
As of September 15, 2011 several companies are pursuing treatments for FM. Chelsea Therapeutics International, Inc. (CHTP) is developing droxidopa for the treatment of fibromyalgia. Droxidopa is a precursor of the neurotransmitter norephinephrine which suggests it would compete with Cymbalta and Savella which also increase norephinephrine activity. Clinical trials in the U.S. are registered with the FDA and reported on the website, www.ClinicalTrials.gov. A trial of Amrix is recruiting subjects (trial NCT01041495), which may indicate that Cephalon is developing its long-acting formulation of cyclobenzaprine to treat symptoms of FM. Another trial of Ultracet® (tramadol and acetaminophen combination) is listed (trial NCT00766675), which may indicate that Johnson and Johnson is developing Ultracet to treat symptoms of FM.
A number of companies are specifically engaged in developing drugs for PTSD. According to ClinicalTrials.gov, ongoing or recent trials of medicines include: quetiapine by AstraZeneca (trial NCT00237393) and by Mclean Hospital (trial NCT01066156), levetiracetam by UCB (trial NCT00413296), Δ9-THC by Hadassah Medical Organization (trial NCT00965809), paroxetine hydrochloride hydrate by GlaxoSmithKline (trial NCT00557622), topiramate by Ortho-McNeil Janssen Scientific Affairs (trial NCT00203463), hydrocortisone by Lightfighter Trust (trial NCT01090518), mirtazapine by Research Foundation for Mental Hygiene (trial NCT01178671) and by Department of Veterans Affairs (trial NCT00302107), orvepitant by GlaxoSmithKline (trial NCT01000493), d-cycloserine by Weill Medical College of Cornell University (trial NCT00875342), duloxetine by Yale University (trial NCT00763178), ziprasidone by Pfizer (trial NCT00208208),and aripiprazole by Durham VA Medical Center (trial NCT00489866). Other medications that may be used for the treatment of PTSD include anti-depressants such as: nefazodone and trazodone; the antihistamine cyproheptadine and certain atypical antipsychotics such as olanzapine and risperidone. Several of these products are supported by companies such as AstraZeneca, GlaxoSmithKline and Pfizer.
A potential competing medication for treating FM symptoms at bedtime had been Rekinla® which was being developed by Jazz Pharmaceuticals, or Jazz. The active ingredient in Rekinla is sodium oxybate, which results in profound sedation and amnesia. Sodium oxybate is the active ingredient in XYREM®, approved by the FDA for the treatment of excessive daytime sleepiness and cataplexy, the sudden loss of muscle tone, in adult patients with narcolepsy. Rekinla is administered at bedtime and a second dose is administered by awakening the patient four hours later. Jazz’ studies of Rekinla showed that a treatment that affects sleep quality can improve FM symptoms to meet FDA requirements for an effective product. While Jazz obtained compelling evidence supporting the efficacy of its treatment on FM symptoms, the FDA rejected their application to market Rekinla for treating FM in 2010. Sodium oxybate is a controlled substance under the auspices of the Drug Enforcement Administration (DEA). In June 2011, Jazz publicly announced their intention to cease development of Rekinla for FM.
| 14 |
Intellectual Property
Proprietary protection for our product candidates, technology and processes are important to our business and we seek patent protection in the U.S. and internationally when we deem appropriate. We also rely on trade secrets, know-how and continuing technological advances to protect various aspects of our core technology. We require our employees, consultants and scientific collaborators to execute confidentiality and invention assignment agreements with us.
We own numerous patents and have patent applications pending in the United States and abroad. In addition, we have one trademark application pending.
Our commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection of our current and future product candidates and the methods used to manufacture them, as well as successfully defending these patents against third-party challenges. Our ability to stop third parties from making, using, selling, offering to sell or importing our products depends on the extent to which we have rights under valid and enforceable patents or trade secrets that cover these activities. We cannot assure you that our pending patent applications will result in issued patents.
Approved Patents
Our current patents owned are as follows:
|
Number |
Name |
Jurisdiction |
Expiration Date | |||
| 6,541,523 | “Methods For Treating Or Preventing Fibromyalgia Using Very Low Doses Of Cyclobenzaprine” | U.S.A. | August 11, 2020 | |||
| 6,395,788 | “Methods And Compositions For Treating Or Preventing Sleep Disturbances And Associated Illnesses Using Very Low Doses Of Cyclobenzaprine” | U.S.A. | August 11, 2020 | |||
| 6,358,944 | “Methods And Compositions For Treating Generalized Anxiety Disorder” | U.S.A. | August 11, 2020 | |||
| EP 1202722 | “Uses of Compositions for Treating or Preventing Sleep Disturbances Using Very Low Doses of Cyclobenzaprine” | European Patent Office, Belgium, France, Ireland, Luxembourg, Monaco, Portugal, Switzerland and United Kingdom | August 11, 2020 | |||
| AT 299369 | “Uses of Compositions for Treating or Preventing Sleep Disturbances Using Very Low Doses of Cyclobenzaprine” | Austria | August 11, 2020 | |||
| DE 60021266 | “Uses of Compositions for Treating or Preventing Sleep Disturbances Using Very Low Doses of Cyclobenzaprine” | Germany | August 11, 2020 | |||
| NZ 516749 | “Uses of Compositions for Treating or Preventing Sleep Disturbances Using Very Low Doses of Cyclobenzaprine” | New Zealand | August 11, 2020 | |||
| ES 2245944 | “Uses of Compositions for Treating or Preventing Sleep Disturbances Using Very Low Doses of Cyclobenzaprine” | Spain | August 11, 2020 | |||
| HK 1047691 | “Uses of Compositions for Treating or Preventing Sleep Disturbances Using Very Low Doses of Cyclobenzaprine” | Hong Kong | August 11, 2020 | |||
| 8,093,300 | “Compositions and Methods for Increasing Compliance with Therapies using Aldehyde Dehydrogenase Inhibitors and Treating Alcoholism” (notice of allowance) | U.S.A. | November 4, 2021 | |||
| AU 2002354017 | “Compositions and Methods for Increasing Compliance with Therapies using Aldehyde Dehydrogenase Inhibitors and Treating Alcoholism” | Australia | November 4, 2022 | |||
| CA 2463987 | “Compositions and Methods for Increasing Compliance with Therapies using Aldehyde Dehydrogenase Inhibitors and Treating Alcoholism” | Canada | November 4, 2022 | |||
| EP 1441708 | “Compositions and Methods for Increasing Compliance with Therapies using Aldehyde Dehydrogenase Inhibitors and Treating Alcoholism” |
European Patent Office, Austria, Belgium, Switzerland, Denmark, Luxembourg, Monaco, Germany, France, Portugal and United Kingdom |
November 4, 2022 | |||
| NZ 532583 | “Compositions and Methods for Increasing Compliance with Therapies using Aldehyde Dehydrogenase Inhibitors and Treating Alcoholism” | New Zealand | November 4, 2022 |
| 15 |
Patent Applications
Our current patent applications that are pending are as follows:
Number |
Name |
Jurisdiction | ||
| 12/948,828 | “Methods And Compositions For Treating Symptoms Associated With Post-Traumatic Stress Disorder Using Cyclobenzaprine” | U.S.A. | ||
| 61/449,838 | “Methods and Compositions for Treating Depression Using Cyclobenzaprine” | U.S.A. | ||
| 13/157,270 | “Method for Improving Fatigue Using Low Dose Cyclobenzaprine” | U.S.A. | ||
| PCT/US 10/02979 | “Methods And Compositions For Treating Symptoms Associated With Post-Traumatic Stress Disorder Using Cyclobenzaprine” | PCT | ||
| PCT/US 11/01529 | “Method for Treating Cocaine Addiction” | PCT | ||
| 12/151,200 | “Method For Treating Neurodegenerative Dysfunction” | U.S.A. | ||
| CA 2723688 | “Method For Treating Neurodegenerative Dysfunction” | Canada | ||
| EP 2299822 | “Method For Treating Neurodegenerative Dysfunction” | European Patent Office |
Trademark Application
We have one trademark application that is pending as follows:
Number |
Name |
Jurisdiction | ||
| 85088881 | Tonix Pharmaceuticals | U.S.A. |
Research and Development
We have one employee dedicated to research and development. We anticipate that our research and development expenditures will increase several fold as we move TNX-102 and TNX-105 into clinical development and investigate other product candidates for incorporation into our portfolio. We need to raise additional capital to fund our development plans and there is no certainty that we will be successful in continuing to attract new investments. Our research and development operations are located in New York, NY. We expect to use third parties to conduct our preclinical and clinical trials.
Manufacturing
We intend to contract with third parties for the manufacture of our compounds for investigational purposes, for preclinical and clinical testing and for any FDA approved products for commercial sale. We have contracted with Lipocine Inc. to manufacture TNX-102 for use in our ongoing pharmacokinetic study. We will need to contract with a larger scale cGMP contract manufacturer for product to be used in further studies of TNX-102, which we do not anticipate any problems in securing as needed. All of our compounds are small molecules, generally constructed using industry standard processes and use readily accessible raw materials.
Government Regulation
The FDA and other federal, state, local and foreign regulatory agencies impose substantial requirements upon the clinical development, approval, labeling, manufacture, marketing and distribution of drug products. These agencies regulate, among other things, research and development activities and the testing, approval, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, advertising and promotion of our product candidates. The regulatory approval process is generally lengthy and expensive, with no guarantee of a positive result. Moreover, failure to comply with applicable FDA or other requirements may result in civil or criminal penalties, recall or seizure of products, injunctive relief including partial or total suspension of production, or withdrawal of a product from the market.
| 16 |
The FDA regulates, among other things, the research, manufacture, promotion and distribution of drugs in the United States under the FFDCA and other statutes and implementing regulations. The process required by the FDA before prescription drug product candidates may be marketed in the United States generally involves the following:
| — | completion of extensive nonclinical laboratory tests, animal studies and formulation studies, all performed in accordance with the FDA’s Good Laboratory Practice regulations; | |
| — | submission to the FDA of an IND, which must become effective before human clinical trials may begin; | |
| — | for some products, performance of adequate and well-controlled human clinical trials in accordance with the FDA’s regulations, including Good Clinical Practices, to establish the safety and efficacy of the product candidate for each proposed indication; | |
| — | submission to the FDA of an NDA; | |
| — | satisfactory completion of an FDA preapproval inspection of the manufacturing facilities at which the product is produced to assess compliance with current Good Manufacturing Practice, or cGMP, regulations; and | |
| — | FDA review and approval of the NDA prior to any commercial marketing, sale or shipment of the drug. |
The testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our product candidates will be granted on a timely basis, if at all.
Nonclinical tests include laboratory evaluations of product chemistry, formulation and stability, as well as studies to evaluate toxicity in animals and other animal studies. The results of nonclinical tests, together with manufacturing information and analytical data, are submitted as part of an IND to the FDA. Some nonclinical testing may continue even after an IND is submitted. The IND also includes one or more protocols for the initial clinical trial or trials and an investigator’s brochure. An IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions relating to the proposed clinical trials as outlined in the IND and places the clinical trial on a clinical hold. In such cases, the IND sponsor and the FDA must resolve any outstanding concerns or questions before any clinical trials can begin. Clinical trial holds also may be imposed at any time before or during studies due to safety concerns or non-compliance with regulatory requirements. An independent institutional review board, or IRB, at each of the clinical centers proposing to conduct the clinical trial must review and approve the plan for any clinical trial before it commences at that center. An IRB considers, among other things, whether the risks to individuals participating in the trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the consent form signed by the trial participants and must monitor the study until completed.
Clinical Trials
Clinical trials involve the administration of the product candidate to human subjects under the supervision of qualified medical investigators according to approved protocols that detail the objectives of the study, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor participant safety. Each protocol is submitted to the FDA as part of the IND.
Human clinical trials are typically conducted in three sequential phases, but the phases may overlap, or be combined.
| — | Phase 1 clinical trials typically involve the initial introduction of the product candidate into healthy human volunteers. In Phase 1 clinical trials, the product candidate is typically tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and pharmacodynamics. | |
| — | Phase 2 clinical trials are conducted in a limited patient population to gather evidence about the efficacy of the product candidate for specific, targeted indications; to determine dosage tolerance and optimal dosage; and to identify possible adverse effects and safety risks. | |
| — | Phase 3 clinical trials are undertaken to evaluate clinical efficacy and to test for safety in an expanded patient population at geographically dispersed clinical trial sites. The size of Phase 3 clinical trials depends upon clinical and statistical considerations for the product candidate and disease, but sometimes can include several thousand patients. Phase 3 clinical trials are intended to establish the overall risk-benefit ratio of the product candidate and provide an adequate basis for product labeling. |
Clinical testing must satisfy extensive FDA regulations. Reports detailing the results of the clinical trials must be submitted at least annually to the FDA and safety reports must be submitted for serious and unexpected adverse events. Success in early stage clinical trials does not assure success in later stage clinical trials. The FDA, an IRB or we may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk.
| 17 |
New Drug Applications
Assuming successful completion of the required clinical trials, the results of product development, nonclinical studies and clinical trials are submitted to the FDA as part of an NDA. An NDA also must contain extensive manufacturing information, as well as proposed labeling for the finished product. An NDA applicant must develop information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing the product in accordance with cGMP. The manufacturing process must be capable of consistently producing quality product within specifications approved by the FDA. The manufacturer must develop methods for testing the quality, purity and potency of the final product. In addition, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product does not undergo unacceptable deterioration over its shelf life. Prior to approval, the FDA will conduct an inspection of the manufacturing facilities to assess compliance with cGMP.
The FDA reviews all NDAs submitted before it accepts them for filing. The FDA may request additional information rather than accept an NDA for filing. In this event, the NDA must be resubmitted with the additional information and is subject to review before the FDA accepts it for filing. After an application is filed, the FDA may refer the NDA to an advisory committee for review, evaluation and recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendation of an advisory committee, but it considers them carefully when making decisions. The FDA may deny approval of an NDA if the applicable regulatory criteria are not satisfied. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. The FDA may issue a complete response letter, which may require additional clinical or other data or impose other conditions that must be met in order to secure final approval of the NDA. If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. In addition, the FDA may require us to conduct Phase 4 testing which involves clinical trials designed to further assess a drug’s safety and effectiveness after NDA approval, and may require surveillance programs to monitor the safety of approved products which have been commercialized. Once issued, the FDA may withdraw product approval if ongoing regulatory requirements are not met or if safety or efficacy questions are raised after the product reaches the market.
Section 505(b)(2) NDAs
There are two types of NDAs: the full NDA and the Section 505(b)(2) NDA. When possible, we intend to file Section 505(b)(2) NDAs that might, if accepted by the FDA, save time and expense in the development and testing of our product candidates. A full NDA is submitted under Section 505(b)(1) of the FFDCA, and must contain full reports of investigations conducted by the applicant to demonstrate the safety and effectiveness of the drug. A Section 505(b)(2) NDA may be submitted for a drug for which one or more of the investigations relied upon by the applicant was not conducted by or for the applicant and for which the applicant has no right of reference from the person by or for whom the investigations were conducted. A Section 505(b)(2) NDA may be submitted based in whole or in part on published literature or on the FDA’s finding of safety and efficacy of one or more previously approved drugs, which are known as reference drugs. Thus, the filing of a Section 505(b)(2) NDA may result in approval of a drug based on fewer clinical or nonclinical studies than would be required under a full NDA. The number and size of studies that need to be conducted by the sponsor depends on the amount and quality of data pertaining to the reference drug that are publicly available, and on the similarity of and differences between the applicant’s drug and the reference drug. In some cases, extensive, time-consuming, and costly clinical and nonclinical studies may still be required for approval of a Section 505(b)(2) NDA.
Because we are developing new formulations of previously approved chemical entities, such as cyclobenzaprine, our drug approval strategy is to submit Section 505(b)(2) NDAs to the FDA. The FDA may not agree that our product candidates are approvable as Section 505(b)(2) NDAs. If the FDA determines that Section 505(b)(2) NDAs are not appropriate and that full NDAs are required for our product candidates, the time and financial resources required to obtain FDA approval for our product candidates could substantially and materially increase, and our products might be less likely to be approved. If the FDA requires full NDAs for our product candidates, or requires more extensive testing and development for some other reason, our ability to compete with alternative products that arrive on the market more quickly than our product candidates would be adversely impacted.
Based on our intent to file under Section 505(b)(2) with respect to our two lead product candidates, we believe it is unlikely the development process for these product candidates will follow the ordinary course of Phase 1, Phase 2 and Phase 3 studies. Our planned human pharmacokinetics study of reformulated cyclobenzaprine pills will represent the first use of TNX-102 in humans and could therefore be described as “Phase 1.” However, because the study will compare TNX-102 to existing approved formulations of cyclobenzaprine and will specify the comparable ability to deliver effective levels of cyclobenzaprine to the bloodstream of FM patients, this study will also provide a reference to the therapeutic effects previously observed in our dose-ranging clinical study of immediate-release cyclobenzaprine tablets in FM patients. For these reasons, rather than always identifying clinical trials by Phase, we find it more illustrative to describe in a narrative form the purpose of the studies and the nature and potential significance of the results. Because our double-blind, randomized, placebo-controlled, dose-ranging study on bedtime cyclobenzaprine was performed in Canada, we have not had meetings with the FDA’s Center for Drug Evaluation and Research to discuss our approach and plans.
| 18 |
Patent Protections
An applicant submitting a Section 505(b)(2) NDA must certify to the FDA with respect to the patent status of the reference drug upon which the applicant relies in support of approval of its drug. With respect to every patent listed in the FDA’s Orange Book, which is the FDA’s list of approved drug products, as claiming the reference drug or an approved method of use of the reference drug, the Section 505(b)(2) applicant must certify that: (1) there is no patent information listed by the FDA for the reference drug; (2) the listed patent has expired; (3) the listed patent has not expired, but will expire on a particular date; (4) the listed patent is invalid, unenforceable, or will not be infringed by the manufacture, use, or sale of the product in the Section 505(b)(2) NDA; or (5) if the patent is a use patent, that the applicant does not seek approval for a use claimed by the patent. If the applicant files a certification to the effect of clause (1), (2) or (5), FDA approval of the Section 505(b)(2) NDA may be made effective immediately upon successful FDA review of the application, in the absence of marketing exclusivity delays, which are discussed below. If the applicant files a certification to the effect of clause (3), the Section 505(b)(2) NDA approval may not be made effective until the expiration of the relevant patent and the expiration of any marketing exclusivity delays.
If the Section 505(b)(2) NDA applicant provides a certification to the effect of clause (4), referred to as a paragraph IV certification, the applicant also must send notice of the certification to the patent owner and the holder of the NDA for the reference drug. The filing of a patent infringement lawsuit within 45 days of the receipt of the notification may prevent the FDA from approving the Section 505(b)(2) NDA for 30 months from the date of the receipt of the notification unless the court determines that a longer or shorter period is appropriate because either party to the action failed to reasonably cooperate in expediting the action. However, the FDA may approve the Section 505(b)(2) NDA before the 30 months have expired if a court decides that the patent is invalid, unenforceable, or not infringed, or if a court enters a settlement order or consent decree stating the patent is invalid or not infringed.
Notwithstanding the approval of many products by the FDA pursuant to Section 505(b)(2), over the last few years certain brand-name pharmaceutical companies and others have objected to the FDA’s interpretation of Section 505(b)(2). If the FDA’s interpretation of Section 505(b)(2) is successfully challenged in court, the FDA may be required to change its interpretation of Section 505(b)(2) which could delay or even prevent the FDA from approving any Section 505(b)(2) NDA that we submit. The pharmaceutical industry is highly competitive, and it is not uncommon for a manufacturer of an approved product to file a citizen petition with the FDA seeking to delay approval of, or impose additional approval requirements for, pending competing products. If successful, such petitions can significantly delay, or even prevent, the approval of the new product. Moreover, even if the FDA ultimately denies such a petition, the FDA may substantially delay approval while it considers and responds to the petition.
Marketing Exclusivity
Market exclusivity provisions under the FFDCA can delay the submission or the approval of Section 505(b)(2) NDAs, thereby delaying a Section 505(b)(2) product from entering the market. The FFDCA provides five-year marketing exclusivity to the first applicant to gain approval of an NDA for a new chemical entity, or NCE, meaning that the FDA has not previously approved any other drug containing the same active moiety. This exclusivity prohibits the submission of a Section 505(b)(2) NDA for any drug product containing the active ingredient during the five-year exclusivity period. However, submission of a Section 505(b)(2) NDA that certifies that a listed patent is invalid, unenforceable, or will not be infringed, as discussed above, is permitted after four years, but if a patent infringement lawsuit is brought within 45 days after such certification, FDA approval of the Section 505(b)(2) NDA may automatically be stayed until 7 1/2 years after the NCE approval date. The FFDCA also provides three years of marketing exclusivity for the approval of new and supplemental NDAs for product changes, including, among other things, new indications, dosage forms, routes of administration or strengths of an existing drug, or for a new use, if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by FDA to be essential to the approval of the application. Five-year and three-year exclusivity will not delay the submission or approval of another full NDA; however, as discussed above, an applicant submitting a full NDA under Section 505(b)(1) would be required to conduct or obtain a right of reference to all of the preclinical and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
Other types of exclusivity in the United States include orphan drug exclusivity and pediatric exclusivity. The FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making available in the United States a drug for this type of disease or condition will be recovered from sales in the United States for that drug. Seven-year orphan drug exclusivity is available to a product that has orphan drug designation and that receives the first FDA approval for the indication for which the drug has such designation. Orphan drug exclusivity prevents approval of another application for the same drug for the same orphan indication, for a period of seven years, regardless of whether the application is a full NDA or a Section 505(b)(2) NDA, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity. Pediatric exclusivity, if granted, provides an additional six months to an existing exclusivity or statutory delay in approval resulting from a patent certification. This six-month exclusivity, which runs from the end of other exclusivity protection or patent delay, may be granted based on the voluntary completion of a pediatric study in accordance with an FDA-issued “Written Request” for such a study.
| 19 |
Section 505(b)(2) NDAs are similar to full NDAs filed under Section 505(b)(1) in that they are entitled to any of these forms of exclusivity if they meet the qualifying criteria. They also are entitled to the patent protections described above, based on patents that are listed in the FDA’s Orange Book in the same manner as patents claiming drugs and uses approved for NDAs submitted as full NDAs.
Other Regulatory Requirements
Maintaining substantial compliance with appropriate federal, state and local statutes and regulations requires the expenditure of substantial time and financial resources. Drug manufacturers are required to register their establishments with the FDA and certain state agencies, and after approval, the FDA and these state agencies conduct periodic unannounced inspections to ensure continued compliance with ongoing regulatory requirements, including cGMPs. In addition, after approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further FDA review and approval. The FDA may require post-approval testing and surveillance programs to monitor safety and the effectiveness of approved products that have been commercialized. Any drug products manufactured or distributed by us pursuant to FDA approvals are subject to continuing regulation by the FDA, including:
| — | record-keeping requirements; | |
| — | reporting of adverse experiences with the drug; | |
| — | providing the FDA with updated safety and efficacy information; | |
| — | reporting on advertisements and promotional labeling; | |
| — | drug sampling and distribution requirements; and | |
| — | complying with electronic record and signature requirements. |
In addition, the FDA strictly regulates labeling, advertising, promotion and other types of information on products that are placed on the market. There are numerous regulations and policies that govern various means for disseminating information to health-care professionals as well as consumers, including to industry sponsored scientific and educational activities, information provided to the media and information provided over the Internet. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label.
The FDA has very broad enforcement authority and the failure to comply with applicable regulatory requirements can result in administrative or judicial sanctions being imposed on us or on the manufacturers and distributors of our approved products, including warning letters, refusals of government contracts, clinical holds, civil penalties, injunctions, restitution, and disgorgement of profits, recall or seizure of products, total or partial suspension of production or distribution, withdrawal of approvals, refusal to approve pending applications, and criminal prosecution resulting in fines and incarceration. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability. In addition, even after regulatory approval is obtained, later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market.
Food and Drug Administration Amendments Act of 2007
In September 2007, the Food and Drug Administration Amendments Act of 2007, or FDAAA, became law. This legislation grants significant new powers to the FDA, many of which are aimed at improving drug safety and assuring the safety of drug products after approval. In particular, the new law authorizes the FDA to, among other things, require post-approval studies and clinical trials, mandate changes to drug labeling to reflect new safety information, and require risk evaluation and mitigation strategies for certain drugs, including certain currently approved drugs. In addition, the new law significantly expands the federal government’s clinical trial registry and results databank and creates new restrictions on the advertising and promotion of drug products. Under the FDAAA, companies that violate these and other provisions of the new law are subject to substantial civil monetary penalties.
The FDA has not yet implemented many of the provisions of the FDAAA, so we cannot predict the impact of the new legislation on the pharmaceutical industry or our business. However, the requirements and changes imposed by the FDAAA may make it more difficult, and more costly, to obtain and maintain approval for new pharmaceutical products, or to produce, market and distribute existing products. In addition, the FDA’s regulations, policies and guidance are often revised or reinterpreted by the agency or the courts in ways that may significantly affect our business and our products. It is impossible to predict whether additional legislative changes will be enacted, or FDA regulations, guidance or interpretations changed, or what the impact of such changes, if any, may be.
| 20 |
Employees
As of March 1, 2012, we had one full time employee, who is Benjamin Selzer. We intend to hire two additional full-time employees in the near future – a Chief Financial Officer and a senior director of drug development/ controller as well as a part-time senior director of research.
In addition, we rely on consultants instead of employees for critical activities, including Seth Lederman who serves as our Chief Executive Officer and as President of Tonix Sub pursuant to a consulting agreement with Lederman & Co., and Seth Lederman and Donald Landry who provide scientific consulting pursuant to a consulting agreement with L&L Technologies, LLC. None of our employees are represented by a labor union, and we believe that our relations with our employees are good.
ITEM 1A - RISK FACTORS
RISKS RELATED TO OUR BUSINESS
We have a history of operating losses and expect to incur losses for the foreseeable future. We may never generate revenues or, if we are able to generate revenues, achieve profitability.
We are focused on product development, and we have not generated any revenues to date. We have incurred losses in each year of our operations, and we expect to continue to incur operating losses for the foreseeable future. These operating losses have adversely affected and are likely to continue to adversely affect our working capital, total assets and shareholders’ equity.
The Company and its prospects should be examined in light of the risks and difficulties frequently encountered by new and early stage companies in new and rapidly evolving markets. These risks include, among other things, the speed at which we can scale up operations, our complete dependence upon development of products that currently have no market acceptance, our ability to establish and expand our brand name, our ability to expand our operations to meet the commercial demand of our clients, our development of and reliance on strategic and customer relationships and our ability to minimize fraud and other security risks.
The process of developing our products requires significant clinical, development and laboratory testing and clinical trials. In addition, commercialization of our product candidates will require that we obtain necessary regulatory approvals and establish sales, marketing and manufacturing capabilities, either through internal hiring or through contractual relationships with others. We expect to incur substantial losses for the foreseeable future as a result of anticipated increases in our research and development costs, including costs associated with conducting preclinical testing and clinical trials, and regulatory compliance activities.
Our ability to generate revenues and achieve profitability will depend on numerous factors, including success in:
| · | developing and testing product candidates; |
| · | receiving regulatory approvals; |
| · | commercializing our products; and |
| · | establishing a favorable competitive position. |
Many of these factors will depend on circumstances beyond our control. We cannot assure you that we will ever have a product approved by the FDA, that we will bring any product to market or, if we are successful in doing so, that we will ever become profitable.
We expect to incur substantial additional operating expenses over the next several years as our research, development, pre-clinical testing, and clinical trial activities increase. The amount of future losses and when, if ever, we will achieve profitability are uncertain. We have no products that have generated any commercial revenue, do not expect to generate revenues from the commercial sale of products in the near future, and might never generate revenues from the sale of products. Our ability to generate revenue and achieve profitability will depend on, among other things, successful completion of the development of our product candidates; obtaining necessary regulatory approvals from the FDA; establishing manufacturing, sales, and marketing arrangements with third parties; and raising sufficient funds to finance our activities. We might not succeed at any of these undertakings. If we are unsuccessful at some or all of these undertakings, our business, prospects, and results of operations may be materially adversely affected.
| 21 |
We received a report from our independent registered public accounting firm with an explanatory paragraph for the year ended December 31, 2011 with respect to our ability to continue as a going concern. The existence of such a report may adversely affect our stock price and our ability to raise capital. There is no assurance that we will not receive a similar report for our year ended December 31, 2012.
In their report dated March 30, 2012, our independent registered public accounting firm expressed substantial doubt about our ability to continue as a going concern as we have incurred losses since inception of development stage, have a negative cash flow from operations and have working capital and stockholders’ deficiencies and require additional financing to fund future operations. Our ability to continue as a going concern is subject to our ability to obtain necessary funding from outside sources, including obtaining additional funding from the sale of our securities, obtaining loans and grants from various financial institutions where possible. Our continued net operating losses increase the difficulty in meeting such goals and there can be no assurances that such methods will prove successful.
We have no approved products on the market and therefore do not expect to generate any revenues from product sales in the foreseeable future, if at all.
To date, we have no approved product on the market and have generated no product revenues. We have funded our operations primarily from sales of our securities. We have not received, and do not expect to receive for at least the next several years, if at all, any revenues from the commercialization of our product candidates. To obtain revenues from sales of our product candidates, we must succeed, either alone or with third parties, in developing, obtaining regulatory approval for, manufacturing and marketing drugs with commercial potential. We may never succeed in these activities, and we may not generate sufficient revenues to continue our business operations or achieve profitability.
We are largely dependent on the success of our lead product candidate, TNX-102, and we cannot be certain that this product candidate will receive regulatory approval or be successfully commercialized.
We currently have no products for sale, and we cannot guarantee that we will ever have any drug products approved for sale. We and our product candidates are subject to extensive regulation by the FDA and comparable regulatory authorities in other countries governing, among other things, research, testing, clinical trials, manufacturing, labeling, promotion, selling, adverse event reporting and recordkeeping. We are not permitted to market any of our product candidates in the United States until we receive approval of an NDA for a product candidate from the FDA or the equivalent approval from a foreign regulatory authority. Obtaining FDA approval is a lengthy, expensive and uncertain process. We currently have one lead product candidate, TNX-102 for the treatment of FM, and the success of our business currently depends on its successful development, approval and commercialization. Any projected sales or future revenue predictions are predicated upon FDA approval and market acceptance of TNX-102. If projected sales do not materialize for any reason, it would have a material adverse effect on our business and our ability to continue operations.
TNX-102 has not completed the clinical development process; therefore, we have not yet submitted an NDA or foreign equivalent or received marketing approval for this product candidate anywhere in the world. The clinical development program for TNX-102 may not lead to commercial products for a number of reasons, including if we fail to obtain necessary approvals from the FDA or foreign regulatory authorities because our clinical trials fail to demonstrate to their satisfaction that this product candidate is safe and effective. We may also fail to obtain the necessary approvals if we have inadequate financial or other resources to advance our product candidates through the clinical trial process. Any failure or delay in completing clinical trials or obtaining regulatory approval for TNX-102 in a timely manner would have a material adverse impact on our business and our stock price.
We need additional capital. If additional capital is not available or is available at unattractive terms, we may be forced to delay, reduce the scope of or eliminate our research and development programs, reduce our commercialization efforts or curtail our operations.
In order to develop and bring our product candidates to market, we must commit substantial resources to costly and time-consuming research, preclinical and clinical trials and marketing activities. We anticipate that our existing cash and cash equivalents will enable us to maintain our current operations for at least the next nine months. We anticipate that we will need an additional $1 million to continue our operations for the next 12 months. We anticipate using our cash and cash equivalents to fund further research and development with respect to our lead product candidates. We may, however, need to raise additional funding sooner if our business or operations change in a manner that consumes available resources more rapidly than we anticipate. Our requirements for additional capital will depend on many factors, including:
| · | successful commercialization of our product candidates; |
| · | the time and costs involved in obtaining regulatory approval for our product candidates; |
| · | costs associated with protecting our intellectual property rights; |
| · | development of marketing and sales capabilities; |
| · | payments received under future collaborative agreements, if any; and |
| · | market acceptance of our products. |
| 22 |
To the extent we raise additional capital through the sale of equity securities, the issuance of those securities could result in dilution to our shareholders. In addition, if we obtain debt financing, a substantial portion of our operating cash flow may be dedicated to the payment of principal and interest on such indebtedness, thus limiting funds available for our business activities. If adequate funds are not available, we may be required to delay, reduce the scope of or eliminate our research and development programs, reduce our commercialization efforts or curtail our operations. In addition, we may be required to obtain funds through arrangements with collaborative partners or others that may require us to relinquish rights to technologies, product candidates or products that we would otherwise seek to develop or commercialize ourselves or license rights to technologies, product candidates or products on terms that are less favorable to us than might otherwise be available.
We will require substantial additional funds to support our research and development activities, and the anticipated costs of preclinical studies and clinical trials, regulatory approvals and eventual commercialization. Such additional sources of financing may not be available on favorable terms, if at all. If we do not succeed in raising additional funds on acceptable terms, we may be unable to initiate clinical trials or obtain approval of any product candidates from the FDA and other regulatory authorities. In addition, we could be forced to discontinue product development, forego sales and marketing efforts and forego attractive business opportunities. Any additional sources of financing will likely involve the issuance of our equity securities, which will have a dilutive effect on our stockholders.
There is no assurance that we will be successful in raising the additional funds needed to fund our business plan. If we are not able to raise sufficient capital in the near future, our continued operations will be in jeopardy and we may be forced to cease operations and sell or otherwise transfer all or substantially all of our remaining assets.
We face intense competition in the markets targeted by our lead product candidates. Many of our competitors have substantially greater resources than we do, and we expect that all of our product candidates under development will face intense competition from existing or future drugs.
We expect that all of our product candidates under development, if approved, will face intense competition from existing and future drugs marketed by large companies. These competitors may successfully market products that compete with our products, successfully identify drug candidates or develop products earlier than we do, or develop products that are more effective, have fewer side effects or cost less than our products.
Additionally, if a competitor receives FDA approval before we do for a drug that is similar to one of our product candidates, FDA approval for our product candidate may be precluded or delayed due to periods of non-patent exclusivity and/or the listing with the FDA by the competitor of patents covering its newly-approved drug product. Periods of non-patent exclusivity for new versions of existing drugs such as our current product candidates can extend up to three and one-half years. See “Business—Government Regulation.”
These competitive factors could require us to conduct substantial new research and development activities to establish new product targets, which would be costly and time consuming. These activities would adversely affect our ability to commercialize products and achieve revenue and profits.
Competition and technological change may make our product candidates and technologies less attractive or obsolete.
We compete with established pharmaceutical and biotechnology companies that are pursuing other forms of treatment for the same indications we are pursuing and that have greater financial and other resources. Other companies may succeed in developing products earlier than us, obtaining FDA approval for products more rapidly, or developing products that are more effective than our product candidates. Research and development by others may render our technology or product candidates obsolete or noncompetitive, or result in treatments or cures superior to any therapy we develop. We face competition from companies that internally develop competing technology or acquire competing technology from universities and other research institutions. As these companies develop their technologies, they may develop competitive positions that may prevent, make futile, or limit our product commercialization efforts, which would result in a decrease in the revenue we would be able to derive from the sale of any products.
There can be no assurance that any of our product candidates will be accepted by the marketplace as readily as these or other competing treatments. Furthermore, if our competitors' products are approved before ours, it could be more difficult for us to obtain approval from the FDA. Even if our products are successfully developed and approved for use by all governing regulatory bodies, there can be no assurance that physicians and patients will accept our product(s) as a treatment of choice.
| 23 |
Furthermore, the pharmaceutical research industry is diverse, complex, and rapidly changing. By its nature, the business risks associated therewith are numerous and significant. The effects of competition, intellectual property disputes, market acceptance, and FDA regulations preclude us from forecasting revenues or income with certainty or even confidence.
If we fail to protect our intellectual property rights, our ability to pursue the development of our technologies and products would be negatively affected.
Our success will depend in part on our ability to obtain patents and maintain adequate protection of our technologies and products. If we do not adequately protect our intellectual property, competitors may be able to use our technologies to produce and market drugs in direct competition with us and erode our competitive advantage. Some foreign countries lack rules and methods for defending intellectual property rights and do not protect proprietary rights to the same extent as the United States. Many companies have had difficulty protecting their proprietary rights in these foreign countries. We may not be able to prevent misappropriation of our proprietary rights.
We have received, and are currently seeking, patent protection for numerous compounds and methods of treating diseases. However, the patent process is subject to numerous risks and uncertainties, and there can be no assurance that we will be successful in protecting our products by obtaining and defending patents. These risks and uncertainties include the following: patents that may be issued or licensed may be challenged, invalidated, or circumvented, or otherwise may not provide any competitive advantage; our competitors, many of which have substantially greater resources than us and many of which have made significant investments in competing technologies, may seek, or may already have obtained, patents that will limit, interfere with, or eliminate our ability to make, use, and sell our potential products either in the United States or in international markets; there may be significant pressure on the United States government and other international governmental bodies to limit the scope of patent protection both inside and outside the United States for treatments that prove successful as a matter of public policy regarding worldwide health concerns; countries other than the United States may have less restrictive patent laws than those upheld by United States courts, allowing foreign competitors the ability to exploit these laws to create, develop, and market competing products.
Moreover, any patents issued to us may not provide us with meaningful protection, or others may challenge, circumvent or narrow our patents. Third parties may also independently develop products similar to our products, duplicate our unpatented products or design around any patents on products we develop. Additionally, extensive time is required for development, testing and regulatory review of a potential product. While extensions of patent term due to regulatory delays may be available, it is possible that, before any of our product candidates can be commercialized, any related patent, even with an extension, may expire or remain in force for only a short period following commercialization, thereby reducing any advantages of the patent.
In addition, the United States Patent and Trademark Office (the “PTO”) and patent offices in other jurisdictions have often required that patent applications concerning pharmaceutical and/or biotechnology-related inventions be limited or narrowed substantially to cover only the specific innovations exemplified in the patent application, thereby limiting the scope of protection against competitive challenges. Thus, even if we or our licensors are able to obtain patents, the patents may be substantially narrower than anticipated.
Our success depends on our patents, patent applications that may be licensed exclusively to us and other patents to which we may obtain assignment or licenses. We may not be aware, however, of all patents, published applications or published literature that may affect our business either by blocking our ability to commercialize our product candidates, by preventing the patentability of our product candidates to us or our licensors, or by covering the same or similar technologies that may invalidate our patents, limit the scope of our future patent claims or adversely affect our ability to market our product candidates.
In addition to patents, we rely on a combination of trade secrets, confidentiality, nondisclosure and other contractual provisions, and security measures to protect our confidential and proprietary information. These measures may not adequately protect our trade secrets or other proprietary information. If they do not adequately protect our rights, third parties could use our technology, and we could lose any competitive advantage we may have. In addition, others may independently develop similar proprietary information or techniques or otherwise gain access to our trade secrets, which could impair any competitive advantage we may have.
Patent protection and other intellectual property protection is crucial to the success of our business and prospects, and there is a substantial risk that such protections will prove inadequate.
| 24 |
We may be involved in lawsuits to protect or enforce our patents, which could be expensive and time consuming.
The pharmaceutical industry has been characterized by extensive litigation regarding patents and other intellectual property rights, and companies have employed intellectual property litigation to gain a competitive advantage. We may become subject to infringement claims or litigation arising out of patents and pending applications of our competitors, or additional interference proceedings declared by the PTO to determine the priority of inventions. The defense and prosecution of intellectual property suits, PTO proceedings, and related legal and administrative proceedings are costly and time-consuming to pursue, and their outcome is uncertain. Litigation may be necessary to enforce our issued patents, to protect our trade secrets and know-how, or to determine the enforceability, scope, and validity of the proprietary rights of others. An adverse determination in litigation or interference proceedings to which we may become a party could subject us to significant liabilities, require us to obtain licenses from third parties, or restrict or prevent us from selling our products in certain markets. Although patent and intellectual property disputes might be settled through licensing or similar arrangements, the costs associated with such arrangements may be substantial and could include our paying large fixed payments and ongoing royalties. Furthermore, the necessary licenses may not be available on satisfactory terms or at all.
Competitors may infringe our patents, and we may file infringement claims to counter infringement or unauthorized use. This can be expensive, particularly for a company of our size, and time-consuming. In addition, in an infringement proceeding, a court may decide that a patent of ours is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover its technology. An adverse determination of any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly.
Also, a third party may assert that our patents are invalid and/or unenforceable. There are no unresolved communications, allegations, complaints or threats of litigation related to the possibility that our patents are invalid or unenforceable. Any litigation or claims against us, whether or not merited, may result in substantial costs, place a significant strain on our financial resources, divert the attention of management and harm our reputation. An adverse decision in litigation could result in inadequate protection for our product candidates and/or reduce the value of any license agreements we have with third parties.
Interference proceedings brought before the U.S. Patent and Trademark Office may be necessary to determine priority of invention with respect to our patents or patent applications. During an interference proceeding, it may be determined that we do not have priority of invention for one or more aspects in our patents or patent applications and could result in the invalidation in part or whole of a patent or could put a patent application at risk of not issuing. Even if successful, an interference proceeding may result in substantial costs and distraction to our management.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation or interference proceedings, there is a risk that some of our confidential information could be compromised by disclosure. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If investors perceive these results to be negative, the price of our common stock could be adversely affected.
If we infringe the rights of third parties we could be prevented from selling products, forced to pay damages, and defend against litigation.
If our products, methods, processes and other technologies infringe the proprietary rights of other parties, we could incur substantial costs and we may have to: obtain licenses, which may not be available on commercially reasonable terms, if at all; abandon an infringing product candidate; redesign our products or processes to avoid infringement; stop using the subject matter claimed in the patents held by others; pay damages; and/or defend litigation or administrative proceedings which may be costly whether we win or lose, and which could result in a substantial diversion of our financial and management resources.
If preclinical testing or clinical trials for our product candidates are unsuccessful or delayed, we will be unable to meet our anticipated development and commercialization timelines.
We rely and expect to continue to rely on third parties, including clinical research organizations and outside consultants, to conduct, supervise or monitor some or all aspects of preclinical testing or clinical trials involving our product candidates. We have less control over the timing and other aspects of these preclinical testing or clinical trials than if we performed the monitoring and supervision entirely on our own. Third parties may not perform their responsibilities for our preclinical testing or clinical trials on our anticipated schedule or, for clinical trials, consistent with a clinical trial protocol. Delays in preclinical and clinical testing could significantly increase our product development costs and delay product commercialization. In addition, many of the factors that may cause, or lead to, a delay in the clinical trials may also ultimately lead to denial of regulatory approval of a product candidate.
The commencement of clinical trials can be delayed for a variety of reasons, including delays in:
| · | demonstrating sufficient safety and efficacy to obtain regulatory approval to commence a clinical trial; |
| · | reaching agreement on acceptable terms with prospective contract research organizations and trial sites; |
| · | manufacturing sufficient quantities of a product candidate; and |
| · | obtaining institutional review board approval to conduct a clinical trial at a prospective site. |
| 25 |
Once a clinical trial has begun, it may be delayed, suspended or terminated by us or the FDA or other regulatory authorities due to a number of factors, including:
| · | ongoing discussions with the FDA or other regulatory authorities regarding the scope or design of our clinical trials; |
| · | failure to conduct clinical trials in accordance with regulatory requirements; |
| · | lower than anticipated recruitment or retention rate of patients in clinical trials; |
| · | inspection of the clinical trial operations or trial sites by the FDA or other regulatory authorities resulting in the imposition of a clinical hold; |
| · | lack of adequate funding to continue clinical trials; |
| · | negative results of clinical trials; or |
| · | side-effects of cyclobenzaprine. |
If clinical trials are unsuccessful, and we are not able to obtain regulatory approvals for our product candidates under development, we will not be able to commercialize these products, and therefore may not be able to generate sufficient revenues to support our business.
If we are unable to file for approval under Section 505(b)(2) of the Federal Food, Drug and Cosmetic Act or if we are required to generate additional data related to safety and efficacy in order to obtain approval under Section 505(b)(2), we may be unable to meet our anticipated development and commercialization timelines.
Our current plans for filing NDAs for our product candidates include efforts to minimize the data we will be required to generate in order to obtain marketing approval for our product candidates and therefore possibly obtain a shortened review period for the applications. While we met with the FDA in August 2011 to discuss initial plans for the future development of TNX-102, we have not yet come to full agreement with the FDA as to the nature or extent of any studies we may be required to conduct in order to achieve approval for any of our product candidates. The timeline for filing and review of our NDAs is based on our plan to submit those NDAs under Section 505(b)(2) of the Federal Food, Drug and Cosmetic Act, wherein we will rely in part on data in the public domain or elsewhere. We have not yet filed an NDA under Section 505(b)(2) for any of our lead product candidates. Depending on the data that may be required by the FDA for approval, some of the data may be related to products already approved by the FDA. If the data relied upon is related to products already approved by the FDA and covered by third-party patents we would be required to certify that we do not infringe the listed patents or that such patents are invalid or unenforceable. As a result of the certification, the third-party would have 45 days from notification of our certification to initiate an action against us. In the event that an action is brought in response to such a certification, the approval of our NDA could be subject to a stay of up to 30 months or more while we defend against such a suit. Approval of our product candidates under Section 505(b)(2) may therefore be delayed until patent exclusivity expires or until we successfully challenge the applicability of those patents to our product candidates. Alternatively, we may elect to generate sufficient additional clinical data so that we no longer rely on data which triggers a potential stay of the approval of our product candidates. Even if no exclusivity periods apply to our applications under Section 505(b)(2), the FDA has broad discretion to require us to generate additional data on the safety and efficacy of our product candidates to supplement third-party data on which we may be permitted to rely. In either event, we could be required, before obtaining marketing approval for any of our product candidates, to conduct substantial new research and development activities beyond those we currently plan to engage in order to obtain approval of our product candidates. Such additional new research and development activities would be costly and time consuming.
We may not be able to obtain shortened review of our applications, and the FDA may not agree that our products qualify for marketing approval. If we are required to generate additional data to support approval, we may be unable to meet our anticipated development and commercialization timelines, may be unable to generate the additional data at a reasonable cost, or at all, and may be unable to obtain marketing approval of our product candidates.
Our executive officers and other key personnel are critical to our business, and our future success depends on our ability to retain them.
Our success depends to a significant extent upon the continued services of Dr. Seth Lederman, our President and Chief Executive Officer. Dr. Lederman has overseen Tonix Sub since inception and provides leadership for our growth and operations strategy as well as being an inventor on many of our patents. Loss of the services of Dr. Lederman would have a material adverse effect on our growth, revenues, and prospective business. We have key-man insurance on the life of Dr. Lederman. We are also highly dependent on the other principal members of our management and scientific team. We are not aware of any present intention of any of our key personnel to leave our company or to retire. However, we have no employment agreement with Dr. Lederman and while we have employment agreements with certain of our employees, all of our employees may terminate their employment at any time. The loss of any of our key personnel, or the inability to attract and retain qualified personnel, may significantly delay or prevent the achievement of our research, development or business objectives and could materially adversely affect our business, financial condition and results of operations.
| 26 |
Any employment agreement we enter into will not ensure the retention of the employee who is a party to the agreement. In addition, we have only limited ability to prevent former employees from competing with us. Furthermore, our future success will also depend in part on the continued service of our key scientific and management personnel and our ability to identify, hire, and retain additional personnel. We experience intense competition for qualified personnel and may be unable to attract and retain the personnel necessary for the development of our business. Moreover, our work force is located in the “Pharmaceutical Corridor” that spans New York, New Jersey and Pennsylvania, where competition for personnel with the scientific and technical skills that we seek is extremely high and is likely to remain high. Because of this competition, our compensation costs may increase significantly.
If we are unable to hire additional qualified personnel, our ability to grow our business may be harmed.
Over time we will need to hire additional qualified personnel with expertise in clinical testing, clinical research and testing, government regulation, formulation and manufacturing, financial matters and sales and marketing. We compete for qualified individuals with numerous biopharmaceutical companies, universities and other research institutions. Competition for such individuals is intense, and we cannot be certain that our search for such personnel will be successful. Attracting and retaining qualified personnel will be critical to our success.
We rely on third parties to manufacture the compounds used in our trials, and we intend to rely on them for the manufacture of any approved products for commercial sale. If these third parties do not manufacture our product candidates in sufficient quantities and at an acceptable cost, clinical development and commercialization of our product candidates could be delayed, prevented or impaired.
We have no manufacturing facilities, and we have no experience in the clinical or commercial-scale manufacture of drugs or in designing drug manufacturing processes. We intend to rely on third parties to manufacture some or all of our product candidates in clinical trials and our products that reach commercialization. Completion of our clinical trials and commercialization of our product candidates requires manufacturing of a sufficient supply of our product candidates. We are currently in discussions with outside sources to manufacture our development compounds. If, for any reason, we become unable to rely on our current sources for the manufacture of our product candidates, either for clinical trials or, at some future date, for commercial quantities, then we would need to identify and contract with additional or replacement third-party manufacturers to manufacture compounds for pre-clinical, clinical, and commercial purposes. We may not be successful in identifying such additional or replacement third-party manufacturers, or in negotiating acceptable terms with any that we do identify.
We believe that there are a variety of manufacturers that we may be able to retain to produce these products. However, once we retain a manufacturing source, if our manufacturers do not perform in a satisfactory manner, we may not be able to develop or commercialize potential products as planned. Certain specialized manufacturers are expected to provide us with modified and unmodified pharmaceutical compounds, including finished products, for use in our preclinical and clinical studies. Some of these materials are available from only one supplier or vendor. Any interruption in or termination of service by such sole source suppliers could result in a delay or interruption in manufacturing until we locate an alternative source of supply. Any delay or interruption in manufacturing operations (or failure to locate a suitable replacement for such suppliers) could materially adversely affect our business, prospects, or results of operations. We do not have any short-term or long-term manufacturing agreements with any of these manufacturers. If we fail to contract for manufacturing on acceptable terms or if third-party manufacturers do not perform as we expect, our development programs could be materially adversely affected. This may result in delays in filing for and receiving FDA approval for one or more of our products. Any such delays could cause our prospects to suffer significantly.
Failure by our third-party manufacturers to comply with the regulatory guidelines set forth by the FDA with respect to our product candidates could delay or prevent the completion of clinical trials, the approval of any product candidates or the commercialization of our products.
Such third-party manufacturers must be inspected by FDA for cGMP compliance before they can produce commercial product. We may be in competition with other companies for access to these manufacturers' facilities and may be subject to delays in manufacture if the manufacturers give other clients higher priority than they give to us. If we are unable to secure and maintain third-party manufacturing capacity, the development and sales of our products and our financial performance may be materially affected.
Manufacturers are obligated to operate in accordance with FDA-mandated requirements. A failure of any of our third-party manufacturers to establish and follow cGMP requirements and to document their adherence to such practices may lead to significant delays in the availability of material for clinical trials, may delay or prevent filing or approval of marketing applications for our products, and may cause delays or interruptions in the availability of our products for commercial distribution following FDA approval. This could result in higher costs to us or deprive us of potential product revenues.
| 27 |
Complying with cGMP and non-U.S. regulatory requirements will require that we expend time, money, and effort in production, recordkeeping, and quality control to assure that the product meets applicable specifications and other requirements. We, or our contracted manufacturing facility, must also pass a pre-approval inspection prior to FDA approval. Failure to pass a pre-approval inspection may significantly delay FDA approval of our products. If we fail to comply with these requirements, we would be subject to possible regulatory action and may be limited in the jurisdictions in which we are permitted to sell our products. As a result, our business, financial condition, and results of operations may be materially harmed.
Drug manufacturers are subject to ongoing periodic unannounced inspections by the FDA, the DEA and corresponding state and foreign agencies to ensure strict compliance with cGMP requirements and other requirements under Federal drug laws, other government regulations and corresponding foreign standards. If we or our third-party manufacturers fail to comply with applicable regulations, sanctions could be imposed on us, including fines, injunctions, civil penalties, failure by the government to grant marketing approval of drugs, delays, suspension or withdrawal of approvals, seizures or recalls of product, operating restrictions and criminal prosecutions.
Corporate and academic collaborators may take actions to delay, prevent, or undermine the success of our products.
Our operating and financial strategy for the development, clinical testing, manufacture, and commercialization of drug candidates is heavily dependent on our entering into collaborations with corporations, academic institutions, licensors, licensees, and other parties. Our current strategy assumes that we will successfully establish these collaborations, or similar relationships; however, there can be no assurance that we will be successful establishing such collaborations. Some of our existing collaborations are, and future collaborations may be, terminable at the sole discretion of the collaborator. Replacement collaborators might not be available on attractive terms, or at all. The activities of any collaborator will not be within our control and may not be within our power to influence. There can be no assurance that any collaborator will perform its obligations to our satisfaction or at all, that we will derive any revenue or profits from such collaborations, or that any collaborator will not compete with us. If any collaboration is not pursued, we may require substantially greater capital to undertake development and marketing of our proposed products and may not be able to develop and market such products effectively, if at all. In addition, a lack of development and marketing collaborations may lead to significant delays in introducing proposed products into certain markets and/or reduced sales of proposed products in such markets.
Data provided by collaborators and others upon which we rely that has not been independently verified could turn out to be false, misleading, or incomplete.
We rely on third-party vendors, scientists, and collaborators to provide us with significant data and other information related to our projects, clinical trials, and our business. If such third parties provide inaccurate, misleading, or incomplete data, our business, prospects, and results of operations could be materially adversely affected.
Our product candidates are novel and still in development.
We are a pharmaceutical company focused on the development of drug product candidates, all of which are still in development. Our drug development methods may not lead to commercially viable drugs for any of several reasons. For example, we may fail to identify appropriate targets or compounds, our drug candidates may fail to be safe and effective in clinical trials, or we may have inadequate financial or other resources to pursue development efforts for our drug candidates. Our drug candidates will require significant additional development, clinical trials, regulatory clearances and additional investment by us or our collaborators before they can be commercialized.
Successful development of our products is uncertain.
Our development of current and future product candidates is subject to the risks of failure and delay inherent in the development of new pharmaceutical products, including: delays in product development, clinical testing, or manufacturing; unplanned expenditures in product development, clinical testing, or manufacturing; failure to receive regulatory approvals; emergence of superior or equivalent products; inability to manufacture on its own, or through any others, product candidates on a commercial scale; and failure to achieve market acceptance.
Because of these risks, our research and development efforts may not result in any commercially viable products. If a significant portion of these development efforts are not successfully completed, required regulatory approvals are not obtained or any approved products are not commercially successfully, our business, financial condition, and results of operations may be materially harmed.
| 28 |
Clinical trials required for our product candidates are expensive and time-consuming, and their outcome is uncertain.
In order to obtain FDA approval to market a new drug product, we must demonstrate proof of safety and effectiveness in humans. To meet these requirements, we must conduct “adequate and well controlled” clinical trials. Conducting clinical trials is a lengthy, time-consuming, and expensive process. The length of time may vary substantially according to the type, complexity, novelty, and intended use of the product candidate, and often can be several years or more per trial. Delays associated with products for which we are directly conducting clinical trials may cause us to incur additional operating expenses. The commencement and rate of completion of clinical trials may be delayed by many factors, including, for example: inability to manufacture sufficient quantities of qualified materials under the FDA's Current Good Manufacturing Practices requirements, commonly known as cGMP, for use in clinical trials; slower than expected rates of patient recruitment; failure to recruit a sufficient number of patients; modification of clinical trial protocols; changes in regulatory requirements for clinical trials; the lack of effectiveness during clinical trials; the emergence of unforeseen safety issues; delays, suspension, or termination of the clinical trials due to the institutional review board responsible for overseeing the study at a particular study site; and government or regulatory delays or “clinical holds” requiring suspension or termination of the trials.
The results from early clinical trials are not necessarily predictive of results obtained in later clinical trials. Accordingly, even if we obtain positive results from early clinical trials, we may not achieve the same success in future clinical trials. Clinical trials may not demonstrate statistically significant safety and effectiveness to obtain the requisite regulatory approvals for product candidates.
Our clinical trials may be conducted in patients with CNS conditions, and in some cases, our product is expected to be used in combination with approved therapies that themselves have significant adverse event profiles. During the course of treatment, these patients could suffer adverse medical events or die for reasons that may or may not be related to our products. We cannot ensure that safety issues will not arise with respect to our products in clinical development.
The failure of clinical trials to demonstrate safety and effectiveness for the desired indications could harm the development of that product candidate and other product candidates. This failure could cause us to abandon a product candidate and could delay development of other product candidates. Any delay in, or termination of, our clinical trials would delay the filing of our NDAs with the FDA and, ultimately, our ability to commercialize our product candidates and generate product revenues. Any change in, or termination of, our clinical trials could materially harm our business, financial condition, and results of operation.
We are subject to extensive and costly government regulation.
Product candidates employing our technology are subject to extensive and rigorous domestic government regulation including regulation by the FDA, the Centers for Medicare and Medicaid Services, other divisions of the United States Department of Health and Human Services, the United States Department of Justice, state and local governments, and their respective foreign equivalents. The FDA regulates the research, development, preclinical and clinical testing, manufacture, safety, effectiveness, record-keeping, reporting, labeling, storage, approval, advertising, promotion, sale, distribution, import, and export of biopharmaceutical products. The FDA regulates small molecule chemical entities as drugs, subject to a New Drug Application, or NDA, under the Federal Food, Drug, and Cosmetic Act. If products employing our technologies are marketed abroad, they will also be subject to extensive regulation by foreign governments, whether or not they have obtained FDA approval for a given product and its uses. Such foreign regulation may be equally or more demanding than corresponding United States regulation.
Government regulation substantially increases the cost and risk of researching, developing, manufacturing, and selling our products. The regulatory review and approval process, which includes preclinical testing and clinical trials of each product candidate, is lengthy, expensive, and uncertain. We or our collaborators must obtain and maintain regulatory authorization to conduct clinical trials. We or our collaborators must obtain regulatory approval for each product we intend to market, and the manufacturing facilities used for the products must be inspected and meet legal requirements. Securing regulatory approval requires the submission of extensive preclinical and clinical data and other supporting information for each proposed therapeutic indication in order to establish the product's safety and efficacy, and in the case of biologics also potency and purity, for each intended use. The development and approval process takes many years, requires substantial resources, and may never lead to the approval of a product.
Even if we are able to obtain regulatory approval for a particular product, the approval may limit the indicated medical uses for the product, may otherwise limit our ability to promote, sell, and distribute the product, may require that we conduct costly post-marketing surveillance, and/or may require that we conduct ongoing post-marketing studies. Material changes to an approved product, such as, for example, manufacturing changes or revised labeling, may require further regulatory review and approval. Once obtained, any approvals may be withdrawn, including, for example, if there is a later discovery of previously unknown problems with the product, such as a previously unknown safety issue.
If we, our collaborators, or our contract manufacturers fail to comply with applicable regulatory requirements at any stage during the regulatory process, such noncompliance could result in, among other things delays in the approval of applications or supplements to approved applications; refusal of a regulatory authority, including the FDA, to review pending market approval applications or supplements to approved applications; warning letters; fines; import and/or export restrictions; product recalls or seizures; injunctions; total or partial suspension of production; civil penalties; withdrawals of previously approved marketing applications or licenses; recommendations by the FDA or other regulatory authorities against governmental contracts; and/or criminal prosecutions.
| 29 |
We do not have, and may never obtain, the regulatory approvals we need to market our product candidates.
Following completion of clinical trials, the results are evaluated and, depending on the outcome, submitted to the FDA in the form of an NDA in order to obtain FDA approval of the product and authorization to commence commercial marketing. In responding to an NDA, the FDA may require additional testing or information, may require that the product labeling be modified, may impose post-approval study or reporting requirements or other restrictions on product distribution, or may deny the application. The FDA has established performance goals for review of NDAs - six months for priority applications and ten months for standard applications. However, the FDA is not required to complete its review within these time periods. The timing of final FDA review and action varies greatly, but can take years in some case and may involve the input of an FDA advisory committee of outside experts. Product sales in the United States may commence only when an NDA is approved.
To date, we have not applied for or received the regulatory approvals required for the commercial sale of any of our products in the United States or in any foreign jurisdiction. None of our product candidates has been determined to be safe and effective, and we have not submitted an NDA to the FDA or an equivalent application to any foreign regulatory authorities for any of our product candidates.
It is possible that none of our product candidates will be approved for marketing. Failure to obtain regulatory approvals, or delays in obtaining regulatory approvals, may adversely affect the successful commercialization of any drugs or biologics that we or our partners develop, may impose additional costs on us or our collaborators, may diminish any competitive advantages that we or our partners may attain, and/or may adversely affect our receipt of revenues or royalties.
Even if approved, our products will be subject to extensive post-approval regulation.
Once a product is approved, numerous post-approval requirements apply. Among other things, the holder of an approved NDA is subject to periodic and other FDA monitoring and reporting obligations, including obligations to monitor and report adverse events and instances of the failure of a product to meet the specifications in the NDA. Application holders must submit new or supplemental applications and obtain FDA approval for certain changes to the approved product, product labeling, or manufacturing process. Application holders must also submit advertising and other promotional material to the FDA and report on ongoing clinical trials.
Depending on the circumstances, failure to meet these post-approval requirements can result in criminal prosecution, fines, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of pre-marketing product approvals, or refusal to allow us to enter into supply contracts, including government contracts. In addition, even if we comply with FDA and other requirements, new information regarding the safety or effectiveness of a product could lead the FDA to modify or withdraw product approval.
Even if we obtain regulatory approval to market our product candidates, our product candidates may not be accepted by the market.
Even if the FDA approves one or more of our product candidates, physicians and patients may not accept it or use it. Even if physicians and patients would like to use our products, our products may not gain market acceptance among healthcare payors such as managed care formularies, insurance companies or government programs such as Medicare or Medicaid. Acceptance and use of our products will depend upon a number of factors including: perceptions by members of the health care community, including physicians, about the safety and effectiveness of our drug or device product; cost-effectiveness of our product relative to competing products; availability of reimbursement for our product from government or other healthcare payers; and effectiveness of marketing and distribution efforts by us and our licensees and distributors, if any.
The degree of market acceptance of any pharmaceutical product that we develop will depend on a number of factors, including:
| · | cost-effectiveness; |
| · | the safety and effectiveness of our products, including any significant potential side effects (including drowsiness and dry mouth), as compared to alternative products or treatment methods; |
| · | the timing of market entry as compared to competitive products; |
| · | flat or declining use of off-label muscle-relaxant products for fibromyalgia prior to the launch of TNX-102; |
| · | the rate of adoption of our products by doctors and nurses; |
| 30 |
| · | product labeling or product insert required by the FDA for each of our products; |
| · | reimbursement policies of government and third-party payors; |
| · | effectiveness of our sales, marketing and distribution capabilities and the effectiveness of such capabilities of our collaborative partners, if any; and |
| · | unfavorable publicity concerning our products or any similar products. |
Our product candidates, if successfully developed, will compete with a number of products manufactured and marketed by major pharmaceutical companies, biotechnology companies and manufacturers of generic drugs. Our products may also compete with new products currently under development by others. Physicians, patients, third-party payors and the medical community may not accept and utilize any of our product candidates. If our products do not achieve market acceptance, we will not be able to generate significant revenues or become profitable.
Because we expect sales of our current product candidates, if approved, to generate substantially all of our product revenues for the foreseeable future, the failure of these products to find market acceptance would harm our business and could require us to seek additional financing.
If we fail to establish marketing, sales and distribution capabilities, or fail to enter into arrangements with third parties, we will not be able to create a market for our product candidates.
Our strategy with our lead product candidates is to control, directly or through contracted third parties, all or most aspects of the product development process, including marketing, sales and distribution. Currently, we do not have any sales, marketing or distribution capabilities. In order to generate sales of any product candidates that receive regulatory approval, we must either acquire or develop an internal marketing and sales force with technical expertise and with supporting distribution capabilities or make arrangements with third parties to perform these services for us. The acquisition or development of a sales and distribution infrastructure would require substantial resources, which may divert the attention of our management and key personnel and defer our product development efforts. To the extent that we enter into marketing and sales arrangements with other companies, our revenues will depend on the efforts of others. These efforts may not be successful. If we fail to develop sales, marketing and distribution channels, or enter into arrangements with third parties, we will experience delays in product sales and incur increased costs.
Sales of pharmaceutical products largely depend on the reimbursement of patients' medical expenses by government health care programs and private health insurers. Without the financial support of the government or third-party payors, the market for our products will be limited. These third-party payors are increasingly challenging the price and examining the cost effectiveness of medical products and services. Recent proposals to change the health care system in the United States have included measures that would limit or eliminate payments for medical products and services or subject the pricing of medical treatment products to government control. Significant uncertainty exists as to the reimbursement status of newly approved health care products. Third-party payors may not reimburse sales of our products or enable our collaborators to sell them at profitable prices.
Our business strategy might involve out-licensing product candidates to or collaborating with larger firms with experience in marketing and selling pharmaceutical products. There can be no assurance that we will be able to successfully establish marketing, sales, or distribution relationships; that such relationships, if established, will be successful; or that we will be successful in gaining market acceptance for our products. To the extent that we enter into any marketing, sales, or distribution arrangements with third parties, our product revenues will be lower than if we marketed and sold our products directly, and any revenues we receive will depend upon the efforts of such third-parties. If we are unable to establish such third-party sales and marketing relationships, or choose not to do so, we will have to establish and rely on our own in-house capabilities.
We, as a company, have no experience in marketing or selling pharmaceutical products and currently have no sales, marketing, or distribution infrastructure. To market any of our products directly, we would need to develop a marketing, sales, and distribution force that both has technical expertise and the ability to support a distribution capability. The establishment of a marketing, sales, and distribution capability would significantly increase our costs, possibly requiring substantial additional capital. In addition, there is intense competition for proficient sales and marketing personnel, and we may not be able to attract individuals who have the qualifications necessary to market, sell, and distribute our products. There can be no assurance that we will be able to establish internal marketing, sales, or distribution capabilities. If we are unable to, or choose not to establish these capabilities, or if the capabilities we establish are not sufficient to meet our needs, we will be required to establish collaborative marketing, sales, or distribution relationships with third parties.
| 31 |
In the event that we are successful in bringing any products to market, our revenues may be adversely affected if we fail to obtain acceptable prices or adequate reimbursement for our products from third-party payors.
Our ability to commercialize pharmaceutical products successfully may depend in part on the availability of reimbursement for our products from:
| · | government and health administration authorities; |
| · | private health insurers; and |
| · | other third party payors, including Medicare. |
We cannot predict the availability of reimbursement for newly-approved health care products. Third-party payors, including Medicare, are challenging the prices charged for medical products and services. Government and other third-party payors increasingly are limiting both coverage and the level of reimbursement for new drugs. Third-party insurance coverage may not be available to patients for any of our products.
The continuing efforts of government and third-party payors to contain or reduce the costs of health care may limit our commercial opportunity. If government and other third-party payors do not provide adequate coverage and reimbursement for any prescription product we bring to market, doctors may not prescribe them or patients may ask to have their physicians prescribe competing drugs with more favorable reimbursement. In some foreign markets, pricing and profitability of prescription pharmaceuticals are subject to government control. In the United States, we expect that there will continue to be federal and state proposals for similar controls. In addition, we expect that increasing emphasis on managed care in the United States will continue to put pressure on the pricing of pharmaceutical products. Cost control initiatives could decrease the price that we receive for any products in the future. Further, cost control initiatives could impair our ability to commercialize our products and our ability to earn revenues from this commercialization.
We face the risk of product liability claims and may not be able to obtain insurance.
Our business exposes us to the risk of product liability claims that are inherent in the development of drugs. If the use of one or more of our or our collaborators' drugs harms people, we may be subject to costly and damaging product liability claims brought against us by clinical trial participants, consumers, health care providers, pharmaceutical companies or others selling our products. Our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of pharmaceutical products we develop, alone or with collaborators. We currently do not carry clinical trial insurance or product liability insurance. We intend to obtain such insurance in the future. We cannot predict all of the possible harms or side effects that may result and, therefore, the amount of insurance coverage we hold now or in the future may not be adequate to cover all liabilities we might incur. We intend to expand our insurance coverage to include the sale of commercial products if we obtain marketing approval for our drug candidates in development, but we may be unable to obtain commercially reasonable product liability insurance for any products approved for marketing. If we are unable to obtain insurance at an acceptable cost or otherwise protect against potential product liability claims, we will be exposed to significant liabilities, which may materially and adversely affect our business and financial position. If we are sued for any injury allegedly caused by our or our collaborators' products, our liability could exceed our total assets and our ability to pay the liability. A product liability claim or series of claims brought against us would decrease our cash and could cause our stock price to fall.
We use hazardous chemicals in our business. Potential claims relating to improper handling, storage or disposal of these chemicals could affect us and be time consuming and costly.
Our research and development processes and/or those of our third party contractors may involve the controlled use of hazardous materials and chemicals. These hazardous chemicals are reagents and solvents typically found in a chemistry laboratory. Our operations also produce hazardous waste products. Federal, state and local laws and regulations govern the use, manufacture, storage, handling and disposal of hazardous materials. While we attempt to comply with all environmental laws and regulations, including those relating to the outsourcing of the disposal of all hazardous chemicals and waste products, we cannot eliminate the risk of contamination from or discharge of hazardous materials and any resultant injury. In the event of such an accident, we could be held liable for any resulting damages and any liability could materially adversely affect our business, financial condition and results of operations.
Compliance with environmental laws and regulations may be expensive. Current or future environmental regulations may impair our research, development or production efforts. We might have to pay civil damages in the event of an improper or unauthorized release of, or exposure of individuals to, hazardous materials. We are not insured against these environmental risks.
| 32 |
If we enter into collaborations with third parties, they might also work with hazardous materials in connection with our collaborations. We may agree to indemnify our collaborators in some circumstances against damages and other liabilities arising out of development activities or products produced in connection with these collaborations.
In addition, the federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous or radioactive materials and waste products may require us to incur substantial compliance costs that could materially adversely affect our business, financial condition and results of operations.
If we retain collaborative partners and our partners do not satisfy their obligations, we will be unable to develop our partnered product candidates.
In the event we enter into any collaborative agreements, we may not have day-to-day control over the activities of our collaborative partners with respect to any of these product candidates. Any collaborative partner may not fulfill its obligations under these agreements. If a collaborative partner fails to fulfill its obligations under an agreement with us, we may be unable to assume the development of the products covered by that agreement or enter into alternative arrangements with a third party. In addition, we may encounter delays in the commercialization of the product candidate that is the subject of the agreement. Accordingly, our ability to receive any revenue from the product candidates covered by these agreements will be dependent on the efforts of our collaborative partner. We could also become involved in disputes with a collaborative partner, which could lead to delays in or termination of our development and commercialization programs and time-consuming and expensive litigation or arbitration. In addition, any such dispute could diminish our collaborators’ commitment to us and reduce the resources they devote to developing and commercializing our products. Conflicts or disputes with our collaborators, and competition from them, could harm our relationships with our other collaborators, restrict our ability to enter future collaboration agreements and delay the research, development or commercialization of our product candidates. If any collaborative partner terminates or breaches its agreement, or otherwise fails to complete its obligations in a timely manner, our chances of successfully developing or commercializing these product candidates would be materially and adversely affected. We may not be able to enter into collaborative agreements with partners on terms favorable to us, or at all. Our inability to enter into collaborative arrangements with collaborative partners, or our failure to maintain such arrangements, would limit the number of product candidates that we could develop and ultimately, decrease our sources of any future revenues.
RISKS RELATED TO OUR STOCK
There has been a limited trading market for our Common Stock and no market activity to date.
Currently, our Common Stock is available for quotation on the Over-the-Counter Bulletin Board under the symbol “TNXP.” However, prior to February 2012, there was no trading activity in our Common Stock and only a few trades have occurred to date. It is anticipated that there will be a limited trading market for the Common Stock on the Over-the-Counter Bulletin Board. The lack of an active market may impair your ability to sell your shares at the time you wish to sell them or at a price that you consider reasonable. The lack of an active market may also reduce the fair market value of your shares. An inactive market may also impair our ability to raise capital by selling shares of capital stock and may impair our ability to acquire other companies or technologies by using Common Stock as consideration.
You may have difficulty trading and obtaining quotations for our Common Stock.
Our Common Stock may not be actively traded, and the bid and asked prices for our Common Stock on the Over-the-Counter Bulletin Board may fluctuate widely. As a result, investors may find it difficult to dispose of, or to obtain accurate quotations of the price of, our securities. This severely limits the liquidity of the Common Stock, and would likely reduce the market price of our Common Stock and hamper our ability to raise additional capital.
The market price for our Common Stock may be volatile, and your investment in our common stock could decline in value.
The stock market in general has experienced extreme price and volume fluctuations. The market prices of the securities of biotechnology and specialty pharmaceutical companies, particularly companies like ours without product revenues and earnings, have been highly volatile and may continue to be highly volatile in the future. This volatility has often been unrelated to the operating performance of particular companies. The following factors, in addition to other risk factors described in this section, may have a significant impact on the market price of our common stock:
| · | announcements of technological innovations or new products by us or our competitors; |
| · | announcement of FDA approval or disapproval of our products or other product-related actions; |
| · | developments involving our discovery efforts and clinical trials; |
| 33 |
| · | developments or disputes concerning patents or proprietary rights, including announcements of infringement, interference or other litigation against us or our potential licensees; |
| · | developments involving our efforts to commercialize our products, including developments impacting the timing of commercialization; |
| · | announcements concerning our competitors, or the biotechnology, pharmaceutical or drug delivery industry in general; |
| · | public concerns as to the safety or efficacy of our products or our competitors’ products; |
| · | changes in government regulation of the pharmaceutical or medical industry; |
| · | changes in the reimbursement policies of third party insurance companies or government agencies; |
| · | actual or anticipated fluctuations in our operating results; |
| · | changes in financial estimates or recommendations by securities analysts; |
| · | developments involving corporate collaborators, if any; |
| · | changes in accounting principles; and |
| · | the loss of any of our key scientific or management personnel. |
In the past, securities class action litigation has often been brought against companies that experience volatility in the market price of their securities. Whether or not meritorious, litigation brought against us could result in substantial costs and a diversion of management’s attention and resources, which could adversely affect our business, operating results and financial condition.
Investor relations activities, nominal “float” and supply and demand factors may affect the price of our stock.
The Company expects to utilize various techniques such as non-deal road shows and investor relations campaigns in order to create investor awareness for the Company. These campaigns may include personal, video and telephone conferences with investors and prospective investors in which our business practices are described. The Company may provide compensation to investor relations firms and pay for newsletters, websites, mailings and email campaigns that are produced by third-parties based upon publicly-available information concerning the Company. The Company will not be responsible for the content of analyst reports and other writings and communications by investor relations firms not authored by the Company or from publicly available information. The Company does not intend to review or approve the content of such analysts’ reports or other materials based upon analysts’ own research or methods. Investor relations firms should generally disclose when they are compensated for their efforts, but whether such disclosure is made or complete is not under our control. In addition, investors in the Company may be willing, from time to time, to encourage investor awareness through similar activities. Investor awareness activities may also be suspended or discontinued which may impact the trading market our common stock.
The SEC and FINRA enforce various statutes and regulations intended to prevent manipulative or deceptive devices in connection with the purchase or sale of any security and carefully scrutinize trading patterns and company news and other communications for false or misleading information, particularly in cases where the hallmarks of “pump and dump” activities may exist, such as rapid share price increases or decreases. We, and our shareholders may be subjected to enhanced regulatory scrutiny due to the small number of holders who initially will own the registered shares of our common stock publicly available for resale, and the limited trading markets in which such shares may be offered or sold which have often been associated with improper activities concerning penny-stocks, such as the OTC Bulletin Board or the OTCQB Marketplace (Pink OTC) or pink sheets. Until such time as our restricted shares are registered or available for resale under Rule 144, there will continue to be a small percentage of shares held by a small number of investors, many of whom acquired such shares in privately negotiated purchase and sale transactions, that will constitute the entire available trading market. The Supreme Court has stated that manipulative action is a term of art connoting intentional or willful conduct designed to deceive or defraud investors by controlling or artificially affecting the price of securities. Often times, manipulation is associated by regulators with forces that upset the supply and demand factors that would normally determine trading prices. Since a small percentage of the outstanding common stock of the Company will initially be available for trading, held by a small number of individuals or entities, the supply of our common stock for sale will be extremely limited for an indeterminate amount of time, which could result in higher bids, asks or sales prices than would otherwise exist. Securities regulators have often cited thinly-traded markets, small numbers of holders, and awareness campaigns as components of their claims of price manipulation and other violations of law when combined with manipulative trading, such as wash sales, matched orders or other manipulative trading timed to coincide with false or touting press releases. There can be no assurance that the Company’s or third-parties’ activities, or the small number of potential sellers or small percentage of stock in the “float,” or determinations by purchasers or holders as to when or under what circumstances or at what prices they may be willing to buy or sell stock will not artificially impact (or would be claimed by regulators to have affected) the normal supply and demand factors that determine the price of the stock.
| 34 |
We do not anticipate paying dividends on our common stock.
We have never declared or paid cash dividends on our common stock and do not expect to do so in the foreseeable future. The declaration of dividends is subject to the discretion of our board of directors and will depend on various factors, including our operating results, financial condition, future prospects and any other factors deemed relevant by our board of directors. You should not rely on an investment in our company if you require dividend income from your investment in our company. The success of your investment will likely depend entirely upon any future appreciation of the market price of our common stock, which is uncertain and unpredictable. There is no guarantee that our common stock will appreciate in value.
We expect that our quarterly results of operations will fluctuate, and this fluctuation could cause our stock price to decline.
Our quarterly operating results are likely to fluctuate in the future. These fluctuations could cause our stock price to decline. The nature of our business involves variable factors, such as the timing of the research, development and regulatory pathways of our product candidates, which could cause our operating results to fluctuate.
Due to the possibility of fluctuations in our revenues and expenses, we believe that quarter-to-quarter comparisons of our operating results are not a good indication of our future performance.
If we or our existing shareholders sell a substantial number of shares of our common stock in the public market, our stock price may decline.
If we or our existing shareholders sell a large number of shares of our common stock, or the public market perceives that we or our existing shareholders might sell shares of common stock, particularly with respect to our affiliates, directors, executive officers or other insiders, the market price of our common stock could decline significantly.
In the future, we may issue additional shares to our employees, directors or consultants, in connection with corporate alliances or acquisitions, or to raise capital. Due to these factors, sales of a substantial number of shares of our common stock in the public market could occur at any time.
Our officers, directors and principal shareholders own a controlling interest in our voting stock and Investors will not have any voice in our management.
Our officers, directors and principal shareholders, in the aggregate, beneficially own or control the votes of approximately 61.26% of our outstanding Common Stock. As a result, these stockholders, acting together, will have the ability to control substantially all matters submitted to our stockholders for approval, including:
| · | election of our board of directors; |
| · | removal of any of our directors; |
| · | amendment of our certificate of incorporation or bylaws; and |
| · | adoption of measures that could delay or prevent a change in control or impede a merger, takeover or other business combination involving us. |
As a result of their ownership and positions, our directors, executive officers and principal shareholders collectively are able to influence all matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions. In addition, sales of significant amounts of shares held by our directors, executive officers or principal shareholders, or the prospect of these sales, could adversely affect the market price of our Common Stock. Management’s stock ownership may discourage a potential acquirer from making a tender offer or otherwise attempting to obtain control of us, which in turn could reduce our stock price or prevent our stockholders from realizing a premium over our stock price.
Our common stock is not currently traded at high volume, and you may be unable to sell at or near ask prices or at all if you need to sell or liquidate a substantial number of shares at one time.
Our common stock is currently traded, but with very low, if any, volume, based on quotations on the “Over-the-Counter Bulletin Board”, meaning that the number of persons interested in purchasing our common stock at or near bid prices at any given time may be relatively small or non-existent. This situation is attributable to a number of factors, including the fact that we are a small company which is still relatively unknown to stock analysts, stock brokers, institutional investors and others in the investment community that generate or influence sales volume, and that even if we came to the attention of such persons, they tend to be risk-averse and would be reluctant to follow an unproven company such as ours or purchase or recommend the purchase of our shares until such time as we became more seasoned and viable. As a consequence, there may be periods of several days or more when trading activity in our shares is minimal or non-existent, as compared to a seasoned issuer which has a large and steady volume of trading activity that will generally support continuous sales without an adverse effect on share price. We cannot give you any assurance that a broader or more active public trading market for our common stock will develop or be sustained, or that trading levels will be sustained.
| 35 |
Shareholders should be aware that, according to Commission Release No. 34-29093, the market for “penny stocks” has suffered in recent years from patterns of fraud and abuse. Such patterns include (1) control of the market for the security by one or a few broker-dealers that are often related to the promoter or issuer; (2) manipulation of prices through prearranged matching of purchases and sales and false and misleading press releases; (3) boiler room practices involving high-pressure sales tactics and unrealistic price projections by inexperienced sales persons; (4) excessive and undisclosed bid-ask differential and markups by selling broker-dealers; and (5) the wholesale dumping of the same securities by promoters and broker-dealers after prices have been manipulated to a desired level, along with the resulting inevitable collapse of those prices and with consequent investor losses. Our management is aware of the abuses that have occurred historically in the penny stock market. Although we do not expect to be in a position to dictate the behavior of the market or of broker-dealers who participate in the market, management will strive within the confines of practical limitations to prevent the described patterns from being established with respect to our securities. The occurrence of these patterns or practices could increase the future volatility of our share price.
Efforts to comply with recently enacted changes in securities laws and regulations will increase our costs and require additional management resources, and we still may fail to comply.
As directed by Section 404 of the Sarbanes-Oxley Act of 2002, the SEC adopted rules requiring public companies to include a report of management on their internal controls over financial reporting in their annual reports on Form 10-K. In addition, in the event we are no longer a smaller reporting company, the independent registered public accounting firm auditing our financial statements would be required to attest to the effectiveness of our internal controls over financial reporting. Such attestation requirement by our independent registered public accounting firm would not be applicable to us until the report for the year ended December 31, 2012 at the earliest, if at all. If we are unable to conclude that we have effective internal controls over financial reporting or if our independent registered public accounting firm is required to, but is unable to provide us with a report as to the effectiveness of our internal controls over financial reporting, investors could lose confidence in the reliability of our financial statements, which could result in a decrease in the value of our securities.
Our common stock is subject to the “penny stock” rules of the SEC and the trading market in our securities is limited, which makes transactions in our stock cumbersome and may reduce the value of an investment in our stock.
The Securities and Exchange Commission (“SEC”) has adopted Rule 15g-9 which establishes the definition of a “penny stock,” for the purposes relevant to us, as any equity security that has a market price of less than $5.00 per share or with an exercise price of less than $5.00 per share, subject to certain exceptions. For any transaction involving a penny stock, unless exempt, the rules require:
| · | that a broker or dealer approve a person’s account for transactions in penny stocks; and | |
| · | the broker or dealer receive from the investor a written agreement to the transaction, setting forth the identity and quantity of the penny stock to be purchased. |
In order to approve a person’s account for transactions in penny stocks, the broker or dealer must:
| · | obtain financial information and investment experience objectives of the person; and | |
| · | make a reasonable determination that the transactions in penny stocks are suitable for that person and the person has sufficient knowledge and experience in financial matters to be capable of evaluating the risks of transactions in penny stocks. |
The broker or dealer must also deliver, prior to any transaction in a penny stock, a disclosure schedule prescribed by the SEC relating to the penny stock market, which, in highlight form:
| · | sets forth the basis on which the broker or dealer made the suitability determination; and | |
| · | that the broker or dealer received a signed, written agreement from the investor prior to the transaction. |
Generally, brokers may be less willing to execute transactions in securities subject to the “penny stock” rules. This may make it more difficult for investors to dispose of our common stock and cause a decline in the market value of our stock.
| 36 |
Disclosure also has to be made about the risks of investing in penny stocks in both public offerings and in secondary trading and about the commissions payable to both the broker-dealer and the registered representative, current quotations for the securities and the rights and remedies available to an investor in cases of fraud in penny stock transactions. Finally, monthly statements have to be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stocks.
FINRA sales practice requirements may also limit a shareholder’s ability to buy and sell our stock.
In addition to the “penny stock” rules described above, FINRA has adopted rules that require that in recommending an investment to a customer, a broker-dealer must have reasonable grounds for believing that the investment is suitable for that customer. Prior to recommending speculative low priced securities to their non-institutional customers, broker-dealers must make reasonable efforts to obtain information about the customer’s financial status, tax status, investment objectives and other information. Under interpretations of these rules, FINRA believes that there is a high probability that speculative low priced securities will not be suitable for at least some customers. The FINRA requirements make it more difficult for broker-dealers to recommend that their customers buy our common stock, which may limit your ability to buy and sell our stock and have an adverse effect on the market for our shares.
ITEM 1B – UNRESOLVED STAFF COMMENTS
Not required under Regulation S-K for “smaller reporting companies.”
ITEM 2 – PROPERTIES
We maintain our principal office at 509 Madison Avenue, Suite 306, New York, New York 10022. Our telephone number at that office is (212) 980-9155 and our fax number is (212) 923-5700. Our current office space consists of approximately 2,355 square feet. The lease expires in September 2015. The base rent is as follows:
| Lease Period | Amount Per Annum | |||
| October 1, 2010 – September 30, 2011 | $ | 120,105.00 | ||
| October 1, 2011 – September 30, 2012 | $ | 123.496.20 | ||
| October 1, 2012 – September 30, 2013 | $ | 126,989.14 | ||
| October 1, 2013 – September 30, 2014 | $ | 130,586.86 | ||
| October 1, 2014 – September 30, 2015 | $ | 134,292.52 | ||
We believe that our existing facilities are suitable and adequate to meet our current business requirements. We maintain websites at www.tonixpharma.com and www.krele.com and the information contained on those websites is not deemed to be a part of this annual report.
ITEM 3 - LEGAL PROCEEDINGS
From time to time, we may become involved in various lawsuits and legal proceedings which arise in the ordinary course of business. However, litigation is subject to inherent uncertainties, and an adverse result in these or other matters may arise from time to time that may harm our business. We are currently not aware of any such legal proceedings or claims that we believe will have, individually or in the aggregate, a material adverse effect on our business, financial condition or operating results.
ITEM 4 – MINE SAFETY DISCLOSURES
Not applicable.
| 37 |
PART II
ITEM 5 - MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Price Range of Common Stock
Our common stock is currently traded on the Over-the-Counter Bulletin Board under the symbol “TNXP.” Prior to October 19, 2011, our common stock was quoted on the Over-the-Counter Bulletin Board under the symbol “TAEI.” Prior to February 2012, no public trades occurred in our common stock.
On March 20, 2012, the closing sale price of our common stock, as reported by the Over-the-Counter Bulletin Board, was $2.00 per share. On March 20, 2012, there were 174 holders of record of our common stock.
Dividend Policy
We have never paid any cash dividends on our capital stock and do not anticipate paying any cash dividends on our common stock in the foreseeable future. We intend to retain future earnings to fund ongoing operations and future capital requirements of our business. Any future determination to pay cash dividends will be at the discretion of the Board and will be dependent upon our financial condition, results of operations, capital requirements and such other factors as the Board deems relevant.
Recent Sales of Unregistered Securities
On October 7, 2011, we issued 22,666,667 shares of our common stock to the shareholders of Tonix Sub in exchange for 100% of the issued and outstanding shares of common stock of Tonix Sub. The shares were issued to accredited investors pursuant to Rule 506 of Regulation D or non-U.S. Persons pursuant to Rule 903 of Regulation S of the Securities Act of 1933, as amended.
On October 7, 2011, we issued 400,000 shares of our common stock to a placement agent in connection with an amendment to a placement agent agreement. The shares were issued to an accredited investor pursuant to Rule 506 of Regulation D or Section 4(2) of the Securities Act of 1933, as amended.
Between October and November 2011, we sold to certain investors (the “Purchasers”) for aggregate cash proceeds of $1,575,000, secured convertible debentures (the “Debentures”) in the principal face amount of $1,575,000 and the exchange of $500,000 in previously issued notes of Tonix Sub that were converted into Debentures in the principal face amount of $500,000 (the “Financing”). The Debentures were sold to accredited investors pursuant to Rule 506 of Regulation D or non-U.S. Persons pursuant to Rule 903 of Regulation S of the Securities Act of 1933, as amended.
The Debentures mature on the earlier of (i) one year from the date of issuance or (ii) the date of closing of a private placement of equity, equity equivalent, convertible debt or debt financing in which we receive gross proceeds, in one or more transactions, of at least $3,425,000 (a “Subsequent Financing”). The Debentures bear interest at 8% per annum and are convertible at the holder’s option into a Subsequent Financing. In the event that a Subsequent Financing has not occurred within 12 months from the date of issuance of the Debenture, the holder has the option to convert the Debenture into a number of shares of our common stock equal to 1% of our shares of common stock on a fully diluted basis for every $125,000 of Debentures (the “Conversion Shares”).
In addition, upon conversion or repayment of the Debenture, the holder is entitled to receive, at the holder’s option, either (i) a warrant (the “Warrant”) to purchase such number of shares of common stock equal to the principal amount of the Debenture divided by the offering price in a Subsequent Financing (the “Warrant Shares”) or (ii) shares of our common stock equal to 33% of the principal amount of the Debenture divided by the offering price in a Subsequent Financing (the “Incentive Shares”).
In connection with the Financing, placement agents earned warrants to purchase shares of Common Stock equal to 3 or 9% of the gross proceeds delivered by Purchasers introduced by such placement agents in the Financing divided by the purchase price per share in the Subsequent Financing (collectively, the “Prior Agent Warrants”). In the event that the Subsequent Financing has not occurred within 12 months from the date of issuance of the Debentures, the placement agents will receive, in lieu of the Prior Agent Warrants, shares of common stock equal to 3 or 9% of the number of shares of Common Stock such Purchasers introduced by such placement agent in the Financing are entitled to receive upon conversion of their Debentures.
| 38 |
ITEM 6 – SELECTED FINANCIAL DATA
Not required under Regulation S-K for “smaller reporting companies.”
ITEM 7 - MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
This Management's Discussion and Analysis of Financial Condition and Results of Operations includes a number of forward-looking statements that reflect Management's current views with respect to future events and financial performance. You can identify these statements by forward-looking words such as “may” “will,” “expect,” “anticipate,” “believe,” “estimate” and “continue,” or similar words. Those statements include statements regarding the intent, belief or current expectations of us and members of its management team as well as the assumptions on which such statements are based. Prospective investors are cautioned that any such forward-looking statements are not guarantees of future performance and involve risk and uncertainties, and that actual results may differ materially from those contemplated by such forward-looking statements.
Readers are urged to carefully review and consider the various disclosures made by us in this report and in our other reports filed with the Securities and Exchange Commission. Important factors currently known to us could cause actual results to differ materially from those in forward-looking statements. We undertake no obligation to update or revise forward-looking statements to reflect changed assumptions, the occurrence of unanticipated events or changes in the future operating results over time. We believe that its assumptions are based upon reasonable data derived from and known about our business and operations and the business and operations of the Company. No assurances are made that actual results of operations or the results of our future activities will not differ materially from its assumptions. Factors that could cause differences include, but are not limited to, expected market demand for the Company’s services, fluctuations in pricing for materials, and competition.
Business Overview
We are a specialty pharmaceutical company focusing on developing new pharmaceuticals products that are safer and more effective than widely prescribed CNS drugs in large and growing markets. The ongoing advances in science and medicine provide a number of opportunities to apply known active pharmaceutical ingredients to new uses. We use the unfolding understanding of disease and medicine when we search for potential therapeutic solutions among prescription pharmaceutical agents that have been used safely in patients for other conditions. We seek to create new dose and formulation options and that are tailored to the new therapeutic uses for these agents.
Many CNS drugs have been identified by physicians who prescribe drugs for some purpose, but observe unexpected improvements in their patients’ CNS conditions. One of our goals is to establish formal clinical study programs to determine if such anecdotal observations are, in fact, reflections of the compound’s ability to treat the CNS condition. While some new applications can use the commercially available form of the drug, in other cases reformulating the active ingredient may improve the active ingredient’s safety or effectiveness in treating the condition. If the formal development programs are proven successful in the clinical tests, we will seek marketing approval from the FDA.
We are currently devoting our efforts to the development of two lead product candidates. Our two most advanced programs are new dose formulations of cyclobenzaprine, which is the active pharmaceutical ingredient of two widely prescribed muscle relaxant products. Due to the well-characterized history of the main active ingredient, we believe our lead products, referred to herein as TNX-102 and TNX-105, have the potential to progress through a shorter development pathway than is typical for drug products based on novel active ingredients. We expect TNX-102 could be approved by FDA after two efficacy studies and a safety exposure study that together would expose the minimum number of FM patients that satisfy FDA’s standards, whereas drug products based on novel active ingredients need exposure to significantly more study subjects.
We also have a pipeline of other product candidates. For commercial reasons, we do not disclose the identities of the active ingredients or targeted indications in our pipeline until a U.S. patent has been allowed or issued. Consistent with our mission, these product candidates are or likely will be reformulations of active ingredients that have been used in humans in other products and which are designed for new CNS therapeutic indications.
In other cases, the products will be formulated to match earlier (“predicate”) products closely enough to be considered generic copies or similar enough to other medications to rely (in part) on their regulatory review and approval. The predicate product may be approved by the FDA under an NDA or may have been reviewed for safety and effectiveness by the National Academy of Sciences under the DESI, in which case they would be considered by FDA to be “unapproved products”. For DESI products, it is our intent to develop NDA versions by modernizing the chemistry, manufacturing and controls and to perform new clinical studies to support an NDA filing.
| 39 |
In August 2010, we formed Krele to commercialize products that are generic versions of predicate NDA products. We anticipate that when our branded products lose patent protection, Krele may market authorized generic versions of them. Krele also may develop or acquire generic products approved under ANDAs and we may market branded versions (branded generics) of such products.
On October 7, 2011, we executed and consummated the Share Exchange Agreement with Tonix Sub. Pursuant to the Share Exchange, each share of Tonix Sub’s common stock was exchanged for 0.9 shares of our common stock, and each share of Tonix Sub’s Series A and B preferred stock was exchanged for 4.8 shares of our common stock. Upon completion of the Share Exchange, the Tonix Sub shareholders, including holders of 1,396,982 restricted shares, which were subject to accelerated vesting, received in exchange for all of their shares, an aggregate of 22,666,667 shares of our common stock and our existing stockholders retained 4,000,000 shares of common stock. The 22,666,667 shares issued to the Tonix Sub shareholders constituted approximately 85% of our 26,666,667 shares of common stock issued and outstanding after the Share Exchange. Upon completion of the Share Exchange, Tonix Sub became our wholly-owned subsidiary. For accounting purposes, the acquisition has been treated as a recapitalization of Tonix Sub, accompanied by the issuance of our common stock for the outstanding common stock of Toxic Sub which was recorded at a nominal value. The historical financial statements are those of Tonix Sub. The accompanying financial statements give retroactive effect to the recapitalization as if it had occurred on June 7, 2007 (inception date). Also, professional services expenses were allocated to research and development and general and administrative expenses in the 2010 and cumulative from inception through December 31, 2011 statement of operations to be consistent with the current period’s presentation.
Current Operating Trends
Our current research and development efforts are focused on developing our lead products, TNX-102 and TNX-105. Our research and development expenses consist of manufacturing studies and the cost of drug ingredients used in such studies, fees paid to providers for conducting various clinical studies as well as for the analysis of the results of such studies and for other medical research addressing the potential efficacy of our drugs. We believe that significant investment in product development is a competitive necessity, and we plan to continue these investments in order to be in a position to realize the potential of our product candidates and proprietary technologies.
We plan to start the next phase of clinical trials for our product candidates TNX-102 and TNX-105 over the next 12 months, subject to raising necessary funds. Clinical trials can be very expensive. If these and additional necessary clinical trials are successful, we plan to prepare and submit applications to the FDA for marketing approval for our drug candidates. This process entails significant costs. As a result of these and other factors, we expect our research and development expenses to increase significantly over the next 12 to 24 months.
We expect that a larger percentage of our research and development expenses in the future will be incurred in support of our current and future preclinical and clinical development programs rather than technology development. These expenditures are subject to numerous uncertainties relating to timing and cost to completion. We test compounds in numerous preclinical studies for safety, toxicology and efficacy. At the appropriate time, subject to the approval of regulatory authorities, we expect to conduct early-stage clinical trials for each drug candidate. We anticipate funding these trials ourselves, and possibly with the assistance of federal grants. As we obtain results from trials, we may elect to discontinue or delay clinical trials for certain products in order to focus our resources on more promising products. Completion of clinical trials may take several years, and the length of time generally varies substantially according to the type, complexity, novelty and intended use of a product candidate.
The commencement and completion of clinical trials for our products may be delayed by many factors, including lack of efficacy during clinical trials, unforeseen safety issues, slower than expected patient recruitment, or government delays. In addition, we may encounter regulatory delays or rejections as a result of many factors, including results that do not support the intended safety or efficacy of our product candidates, perceived defects in the design of clinical trials and changes in regulatory policy during the period of product development. As a result of these risks and uncertainties, we are unable to accurately estimate the specific timing and costs of our clinical development programs or the timing of material cash inflows, if any, from our product candidates. Our business, financial condition and results of operations may be materially adversely affected by any delays in, or termination of, our clinical trials or a determination by the FDA that the results of our trials are inadequate to justify regulatory approval, insofar as cash in-flows from the relevant drug or program would be delayed or would not occur.
| 40 |
Results of Operations
We anticipate that our results of operations will fluctuate for the foreseeable future due to several factors, such as the progress of our research and development efforts and the timing and outcome of regulatory submissions. Due to these uncertainties, accurate predictions of future operations are difficult or impossible to make.
Fiscal year Ended December 31, 2011 Compared to Fiscal year Ended December 31, 2010
Revenues and Cost of Goods Sold. We had no revenues or cost of goods sold during the fiscal years ended December 31, 2011 and 2010.
Research and Development Expenses. Research and development expenses for the fiscal year ended December 31, 2011 were $1,158,167, an increase of $573,869, or 98.2%, from $584,298 for the fiscal year ended December 31, 2010. In 2011, we incurred $342,398 and $318,616 in clinical cost and activities, respectively, as compared to $0 in 2010 for both.
General and Administrative Expenses. General and administrative expenses for the fiscal year ended December 31, 2011 were $2,220,361, an increase of $875,971, or 65%, from $1,344,390 incurred in the fiscal year ended December 31, 2010. This increase is primarily due to payroll related expenses and professional services.
Payroll related expenses increased to $731,284 in the current year from $413,954 for the fiscal year ended December 31, 2010, an increase of $317,330, or 77%. Payroll related expenses include both cash and non-cash compensation associated with the vesting of restricted stock grants. The increase in payroll related costs was a result of a full year of payments to our members of the core management team who joined in June through August of 2010, along with the acceleration of vesting in conjunction with our reverse merger in 2011 of restricted stock previously issued to our employees.
Professional services for the fiscal year ended December 31, 2011 totaled $1,121,547, an increase of $440,642, or 65%, over the $680,905 recognized for the fiscal year ended December 31, 2010. Of professional services, legal fees totaled $373,075 for the fiscal year ended December 31, 2011, an increase of $15,657, or 4.4%, from $357,418 incurred for the fiscal year ended December 31, 2010. Consulting fees totaled $299,144 for the fiscal year ended December 31, 2011, an increase of $139,903 or 87.9%, from $159,241 for the fiscal year ended December 31, 2010. The increase was primarily a result of $189,691 in regulatory costs in the fiscal year ended December 31, 2011 compared to $0 in 2010, offset by a reduction in public relations expenses of $45,513. Accounting fees incurred in fiscal 2011 amounted to $243,003, an increase of $145,400, or 149%, from $97,603 incurred in fiscal 2010. The increase included costs associated with the audit of Tonix Sub’s financial statements for the year ended December 31, 2010, review of our interim financial statements and filings with the SEC related to our recent reverse merger, completed in October 2011.
Travel, meals and entertainment costs for fiscal 2011 were $69,268, an increase of $34,548, or 100%, from $34,720 incurred in fiscal 2010. Travel, meals and entertainment costs include travel related to medical and life sciences conferences. Rent for fiscal 2011 totaled $128,228, an increase of $85,657, or 201%, from $42,571 incurred in fiscal 2010, due primarily to the opening of new office space in New York. Depreciation expense in fiscal 2011 totaled $9,300, an increase of $5,446, or 141%, over the expense of $3,854 incurred in fiscal 2010, as a result of the purchase of new office computers.
Interest Expense. Interest expense for the fiscal year ended December 31, 2011 totaled $91,585, an increase of $55,803, or 156%, from $35,782 incurred during the fiscal year ended December 31, 2010. In 2011, our interest costs were comprised primarily of amortization of deferred financing costs in conjunction with the issuance of our secured convertible debentures in October 2011. We incurred an aggregate of $249,543 in deferred financing costs, of which we amortized $53,377 as interest expense for the fiscal year ended December 31, 2011. In addition, we incurred interest expense related to $500,000 of notes payable and our secured convertible debentures.
Net Loss. As a result of the foregoing, net loss for the year ended December 31, 2011 was $3,470,113, compared to a net loss of $1,964,470 for the year ended December 31, 2010.
Liquidity and Capital Resources
As of December 31, 2011, we had a working capital deficit of $781,723. For the year ended December 31, 2011, we used $2,637,538 of cash in operating activities. Cash provided by financing activities totaled $2,613,000 from the sale of shares of capital stock of $612,000, issuance of notes payable of $500,000 and net proceeds of $1,501,000 from issuance of secured convertible debentures. In the comparable 2010 period, $1,342,000 was raised through the sale of shares of capital stock and $50,000 through the issuance of demand notes, which were converted into shares of capital stock in July 2010. At December 31, 2011, we had cash of $41,123 compared to $65,359 at December 31, 2010. Our cash is held in bank deposit accounts. At December 31, 2011, we had $2,075,000 of secured convertible debentures outstanding.
| 41 |
Cash used in operations for the years ended December 31, 2011 and 2010 was $2,637,538, and $1,233,341, respectively, which represent cash outlays for research and development and general and administrative expenses in such years. Increase in cash outlays principally resulted from clinical cost and activities, regulatory cost, payroll and rent.
Cash generated by investing activities for the year ended December 31, 2011 was $302, compared to a usage of $94,366 in 2010. In 2011, we received a return of a security deposit from space vacated in New Jersey and in 2010 we placed $60,000 in a restricted cash account as collateral for the lease on the New York office. In 2011 and 2010, we purchased office furniture and computer equipment of $2,764 and $34,279, respectively.
In their report dated March 30, 2012, our independent registered public accounting firm stated that our financial statements for the year ended December 31, 2011 were prepared assuming that we would continue as a going concern. Our ability to continue as a going concern is an issue raised due to our net losses and negative cash flows from operations since inception and our expectation that these conditions will continue for the foreseeable future. In addition, we have both working capital and stockholders' deficiencies at December 31, 2011 and require additional financing to fund future operations. Further, we do not have any commercial products available for sale and have not generated revenues and there is no assurance that if approval of our products is received that we will be able to generate cash flow to fund operations. In addition, there can be no assurance that our research and development will be successfully completed or that any product will be approved or commercially viable. Our ability to continue as a going concern is subject to our ability to obtain necessary funding from outside sources, including obtaining additional funding from the sale of our securities, obtaining loans from various financial institutions or being awarded grants from government agencies, where possible. Our continued net operating losses increase the difficulty in meeting such goals and there can be no assurances that such methods will prove successful.
We expect to incur losses from operations for the foreseeable future. We expect to incur increasing research and development expenses, including expenses related to additional clinical trials. We expect that our general and administrative expenses will increase in the future as we expand our business development, add infrastructure and incur additional costs related to being a public company, including incremental audit fees, investor relations programs and increased professional services.
Our future capital requirements will depend on a number of factors, including the progress of our research and development of product candidates, the timing and outcome of regulatory approvals, the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims and other intellectual property rights, the status of competitive products, the availability of financing and our success in developing markets for our product candidates. We believe our existing cash, together with the net proceeds of the 2012 Financing, will be sufficient to fund our operating expenses and capital equipment requirements for the next six months.
We presently do not have any available credit, bank financing or other external sources of liquidity. Due to our history and historical operating losses, our operations have not been a source of liquidity. We will need to obtain additional capital in order to expand operations and become profitable. Future financing may include the issuance of equity or debt securities, obtaining credit facilities, or other financing mechanisms. Even if we are able to raise the funds required, it is possible that we could incur unexpected costs and expenses, fail to collect significant amounts owed to us, or experience unexpected cash requirements that would force us to seek alternative financing. Furthermore, if we issue additional equity or debt securities, stockholders may experience additional dilution or the new equity securities may have rights, preferences or privileges senior to those of existing holders of our common stock.
If additional financing is not available or is not available on acceptable terms, we may be required to delay, reduce the scope of or eliminate our research and development programs, reduce our commercialization efforts or obtain funds through arrangements with collaborative partners or others that may require us to relinquish rights to certain product candidates that we might otherwise seek to develop or commercialize independently.
Notes Pqyable
On September 9, 2011, Tonix Sub sold $500,000 principal amount of convertible notes (the “Notes”) to nine accredited investors. The Notes were due one year from the date of issuance, bear interest at the rate of 8% per annum and were automatically converted into Debentures in the Financing.
| 42 |
2011 Private Placement
Between October and November, 2011 we consummated the Financing pursuant to which we sold $2,075,000 principal amount of Debentures for aggregate cash proceeds of $1,575,000 and the exchange of $500,000 in previously issued Notes of Tonix Sub that were converted into Debentures in the principal face amount of $500,000.
The Debentures mature on the earlier of (i) the one year anniversary of the date of issuance or (ii) the date of closing of a Subsequent Financing. The Debentures bear interest at 8% per annum and are convertible at the holder’s option into a Subsequent Financing. In the event that a Subsequent Financing has not occurred within 12 months from the date of issuance of the Debenture, the holder has the option to convert the Debenture into the Conversion Shares. In addition, upon conversion or repayment of the Debenture, the holders were entitled to receive, at the holder’s option, either (i) the Conversion Warrant or (ii) the Incentive Shares. The private placement that closed in January 2012 met the requirements of a Subsequent Financing, therefore, the holders of the Debentures elected to receive 275,000 Conversion Warrants and 594,000 Incentive Shares. The Conversion Warrants have three year term and $1.00 exercise price.
In connection with the Financing, we made cash payments to WFG Investments and Seagate of $40,000 and $14,000, respectively, as commissions and attorney fees of $20,000. In addition, WFG Investments and Seagate earned an aggregate of 30,750 Prior Agent Warrants, which have terms similar to the Conversion Warrants.
2012 Private Placement
Between January and March, 2012, we consummated a private placement financing transaction (the “2012 Financing”) pursuant to which we issued an aggregate of 264.7106 units (“Units”) to certain investors for aggregate cash proceeds of $4,692,765 and the exchange of $1,925,000 in previously issued Debentures that were converted into Units. The 2012 Financing satisfied the requirements for the Subsequent Financing discussed above.
Each Unit had a purchase price of $25,000 per Unit and consisted of twenty five thousand (25,000) shares of our Common Stock, a Class A Warrant to purchase twenty five thousand (25,000) shares of common stock (the “Class A Warrants”), and a Class B Warrant to purchase up to twenty five thousand (25,000) shares of common stock (the “Class B Warrants” and together with the Class A Warrants, the “Warrants”).
The Class A Warrants have an exercise price of $1.25 per share of Common Stock and will be exercisable for a period of five years from the date of issuance. The Class B Warrants may not be exercised by the Purchasers and will be exercised automatically on their expiration date by cashless exercise or expire without exercise. In the event that the average of our daily volume weighted average price is below $0.75 during the 10 trading days after the Announcement Date (as hereinafter defined) (the “Measuring Period”), then the holder will be entitled to receive additional shares of our Common Stock upon the exercise of the Class B Warrants on the expiration date, which is the 12th trading day after the Announcement Date. In the event that our average daily volume weighted average price is at or above $0.75 during the Measuring Period, the Class B Warrants will expire unexercised. The Announcement Date is the earlier of (1) the date on which we announce via press release the results of the pharmacokinetic study of our TNX-102 drug formulation; or (2) June 1, 2012.
The number of shares issuable upon the cashless exercise of the Class B Warrant is equal to the quotient obtained by dividing [(A-B) [((C-A) *D)/ A]] by (A), where:
(A) = the average of the Company’s daily volume weighted average price during the Measuring Period;
(B) = $0.01, which is the exercise price of the Class B Warrant;
(C) = $1.00, which is the purchase price of the Class B Warrant; and
(D) = the number of shares of common stock purchased by the Class B Warrant holder.
However, for purposes of this calculation, in no event shall the average of our daily volume weighted average price be less than $0.50. For example, in the event that an investor purchases one Unit and the average of our daily volume weighted average price is $0.50, then the Class B Warrant will be exercised and the holder will receive 24,500 shares of Common Stock.
In connection with the Financing, we paid Dawson James Securities, Inc., a FINRA registered broker-dealer (“Dawson James”) a cash payment of $466,777, which represented an 8% commission and a 2% non-accountable expense allowance of the gross proceeds delivered by investors in the 2012 Financing. In addition, Dawson James earned warrants to purchase 466,777 shares of Common Stock, which have an exercise price of $1.25 per share of common stock, will be exercisable for a period of seven years, contain customary anti-dilution protection and are entitled to piggy-back registration rights.
| 43 |
Other
In August 2011, we authorized the initiation of formulation work and manufacturing of TNX-102 for clinical trials pursuant to a contract with Lipocine with respect to a research and development project for reformulation work on our leading products for a fee of $235,000, with work started in the third quarter of 2011. In July 2011, we entered into a contract with a contract research organization in July 2011 to investigate the feasibility of developing a new, proprietary formulation of cyclobenzaprine at a cost of $58,080. In September 2011, we entered into a contract with Pharmanet Canada for contract research work with respect to the pharmacokinetic study for TNX-102. The full cost of the work to be performed is $637,231. Payment is due in four installments based on the achievement of certain performance milestones. In October 2011, we entered into an agreement with another contract research organization to develop, and perform an exploratory pharmacokinetic study on, a new formulation of cyclobenzaprine for an approximate cost of $180,000. In December 2011, we entered into an agreement with a public relations firm to provide news media placement and political intelligence from January 2012 through June 2012 for a total cost of $60,000.
Transactions with Related Parties
Dr. Seth Lederman, our Chief Executive Officer and Chairman of the Board, and Dr. Donald Landry, one of our directors, are the primary founders of Tonix Sub. We have entered into various transactions with several companies under their control, including L&L Technologies, Plumbline, Targent Pharmaceuticals, LLC and Lederman & Co. In 2010, we entered into two-year consulting agreements with L&L Technologies for scientific and medical consulting services, and Lederman & Co. for clinical development, strategic, management and operational consulting services. In consideration for its services, L&L Technologies receives $96,000 per annum. The consulting agreement renews automatically for subsequent terms of one year at $96,000 per annum. Lederman & Co. received $250,000 per annum for its services, until August 1, 2011, when it received $127,000 per annum until such time as we closed on the 2012 Financing. We first closed on the 2012 Financing in January 2012, and effective February 1, 2012, Lederman & Co. receives $250,000 per annum for its services. The consulting agreement renews automatically for subsequent terms of one year at $250,000 per annum. Additionally, on September 9, 2011, L&L Technologies, Targent Pharmaceuticals, LLC and Lederman & Co. purchased $265,000 principal amount of convertible Notes. Such notes were converted into Debentures on October 7, 2011 and in January 2012 such Debentures were converted into the 2012 Financing.
Stock Compensation
In 2010, Tonix Sub’s board of directors and stockholders approved the terms and provisions of the 2010 Stock Plan ("2010 Plan") whereby it reserved 4,564,641 shares of its common stock for issuance pursuant to the 2010 Plan. The 2010 Plan allowed for grants of options to purchase shares of common stock and awards of restricted common stock to employees, officers, directors, consultants and advisors of Tonix Sub. Tonix Sub has granted under the 2010 Plan 2,898,689 shares of restricted stock, 349,082 of which were forfeited and the remaining 2,549,607 shares vested prior to or on October 7, 2011, the date of Share Exchange after which the 2010 Plan ceased to exist. Our stock based compensation expenses amounted to $435,651 in 2011 and $139,882 in 2010.
In February 2012, we approved the 2012 Incentive Stock Options Plan (“2012 Plan”). The 2012 Plan provides for the issuance of options to purchase up to 4,000,000 shares of our common stock to officers, directors, employees and consultants. Under the terms of the 2012 Plan, we may issue Incentive Stock Options, as defined by the Internal Revenue Code, and nonstatutory options. The Board of Directors determines the exercise price, vesting and expiration period of the options granted under the 2012 Plan. However, the exercise price of an Incentive Stock Option must not at least 100% of fair value of the common stock at the date of the grant (or 110% for any stockholder that owns 10% or more of our common stock). The fair market value of the common stock determined based on quoted market price or in absence of such quoted market price, by the Board of Directors in a good faith. Additionally, the vesting period of the grants under the 2012 Plan should not be more than five years and expiration period not more than ten years. We reserved 4,000,000 shares of our common stock for future issuance under the terms of the 2012 Plan.
Lease Commitments
In September 2010, we entered into a five-year lease for office space in New York City, with monthly payments escalating from approximately $10,000 in first year to approximately $11,000 in fifth year. The Company received a rent credit of $9,420 in each of the months of November 2010, December 2010 and January 2011. We issued a letter of credit in the amount of approximately $60,000 for the benefit of the landlord which is collateralized by a money market account. Our future minimum lease payments under the operating lease are as follows:
| Year Ending December 31, | |||||
| 2012 | 124,370 | ||||
| 2013 | 127,889 | ||||
| 2014 | 131,513 | ||||
| 2015 | 100,719 | ||||
| $ | 484,491 | ||||
| 44 |
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses. We evaluate our estimates and judgments on an ongoing basis. We base our estimates on historical experience and on assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates.
We believe the following critical accounting policies affect our more significant judgments and estimates used in the preparation of our consolidated financial statements.
Research and Development. Tonix outsources its research and development efforts and expenses related costs as incurred, including the cost of manufacturing product for testing, licensing fees and costs associated with planning and conducting clinical trials. The value ascribed to patents and other intellectual property acquired was expensed as research and development costs, as it related to particular research and development projects and had no alternative future uses.
Stock Based Compensation. All stock-based payments to employees and to nonemployee directors for their services as directors consisted of grants of restricted stock, which are measured at fair value on the grant date and recognized in the consolidated statements of operations as compensation expense over the relevant vesting period. Restricted stock payments to nonemployees are recognized as an expense over the period of performance. Such payments are measured at fair value at the earlier of the date a performance commitment is reached or the date performance is completed. In addition, for awards that vest immediately and are nonforfeitable the measurement date is the date the award is issued. Because shares of our common stock have not been publicly traded prior to October 7, 2011, we have valued our stock by considering events that have occurred since the date of grants, transactions involving the sale of our common stock to independent third parties and the results of a third party valuation of the projected discounted cash flows of the Company.
Income Taxes. Deferred income tax assets and liabilities are determined based on the estimated future tax effects of net operating loss and credit carryforwards and temporary differences between the tax basis of assets and liabilities and their respective financial reporting amounts measured at the current enacted tax rates. The Company records an estimated valuation allowance on its deferred income tax assets if it is not more likely than not that these deferred income tax assets will be realized. The Company recognizes a tax benefit from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by taxing authorities, based on the technical merits of the position. The tax benefits recognized in the consolidated financial statements from such a position are measured based on the largest benefit that has a greater than 50% likelihood of being realized upon ultimate settlement.
Recent Accounting Pronouncements
There were various updates recently issued, most of which represented technical corrections to the accounting literature or application to specific industries and are not expected to a have a material impact on the Company's consolidated financial position, results of operations or cash flows.
ITEM 7A – QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Not required under Regulation S-K for “smaller reporting companies.”
| 45 |
ITEM 8 - FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
TONIX PHARMACEUTICALS HOLDING CORP.
(Formerly known as Tamandare Explorations, Inc.)
(a development stage company)
| Report of Independent Registered Public Accounting Firm | F-2 | |
| Consolidated balance sheets as of December 31, 2011 and 2010 | F-3 | |
| Consolidated statements of operations for the years ended December 31, 2011 and 2010 and for the period from June 7, 2007 (date of inception) through December 31, 2011 | F-4 | |
| Consolidated statements of stockholders’ deficiency for the years ended December 31, 2011, 2010, 2009, 2008 and for the period from June 7, 2007 (date of inception) through December 31, 2007 | F-5 | |
| Consolidated statements of cash flows for the years ended December 31, 2011 and 2010 and for the period from June 7, 2007 (date of inception) through December 31, 2011 | F-6 | |
| Notes to consolidated financial statements | F-7 – F-17 |
| F-1 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of Directors and Stockholders
Tonix Pharmaceuticals Holding Corp.
We have audited the accompanying consolidated balance sheets of Tonix Pharmaceuticals Holding Corp. (a development stage company) (the “Company”) as of December 31, 2011 and 2010, the related consolidated statements of operations and cash flows for the years than ended and for the period from June 7, 2007 (inception) through December 31, 2011 and the consolidated statements of stockholders’ deficiency for each of the four years in the period ended December 31, 2011 and for the period from June 7, 2007 (inception) through December 31, 2007. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audits include consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Tonix Pharmaceuticals Holding Corp. as of December 31, 2011 and 2010, the consolidated results of its operations and its cash flows for the years ended December 31, 2011 and 2010 and for the period from June 7, 2007 (inception) through December 31, 2011 and consolidated changes in stockholders’ deficiency for each of the four years in the period ended December 31, 2011 and for the period June 7, 2007 through December 31, 2007, in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has incurred recurring net losses and negative cash flows from operations, has both working capital and stockholders' deficiencies at December 31, 2011 and requires additional financing to fund future operations. These events and conditions, among others referred to in Note 2, raise substantial doubt about the Company's ability to continue as a going concern. Management's plans concerning these matters are also described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
| /s/EisnerAmper LLP | |
| EisnerAmper LLP | |
| New York, New York | |
| March 30, 2012 |
| F-2 |
TONIX PHARMACEUTICALS HOLDING CORP.
(Formerly known as Tamandare Explorations Inc.)
(a development stage company)
CONSOLIDATED BALANCE SHEETS
DECEMBER 31, 2011 AND 2010
| 2011 | 2010 | |||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash | $ | 41,123 | $ | 65,359 | ||||
| Prepaid expenses | 102,430 | 23,313 | ||||||
| Total current assets | 143,553 | 88,672 | ||||||
| Furniture and equipment, net | 25,550 | 32,086 | ||||||
| Deferred financing costs, net | 196,166 | - | ||||||
| Restricted cash | 60,177 | 60,087 | ||||||
| Security deposit | - | 3,156 | ||||||
| $ | 425,446 | $ | 184,001 | |||||
| LIABILITIES AND STOCKHOLDERS' DEFICIENCY | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 695,198 | $ | 317,745 | ||||
| Accrued expenses | 10,229 | 22,533 | ||||||
| Accrued interest, including $5,006 to related parties | 38,306 | - | ||||||
| Liability to placement agent | 31,543 | - | ||||||
| Convertible Debentures | 150,000 | - | ||||||
| Total current liabilities | 925,276 | 340,278 | ||||||
| Convertible Debentures, including $265,000 to related parties | 1,925,000 | - | ||||||
| Deferred rent payable | 29,083 | 19,174 | ||||||
| Total liabilities | 2,879,359 | 359,452 | ||||||
| Commitments (Note 6) | - | - | ||||||
| Stockholders' deficiency: | ||||||||
| Common stock, $0.001 par value; 75,000,000 shares authorized, 27,066,667 and 18,034,483 shares issued and outstanding as of December 31, 2011 and 2010, respectively | 27,067 | 18,035 | ||||||
| Additional paid in capital | 3,913,700 | 2,731,081 | ||||||
| Deficit accumulated during development stage | (6,394,680 | ) | (2,924,567 | ) | ||||
| Total stockholders' deficiency | (2,453,913 | ) | (175,451 | ) | ||||
| $ | 425,446 | $ | 184,001 | |||||
See notes to consolidated financial statements
| F-3 |
TONIX PHARMACEUTICALS HOLDING CORP.
(Formerly known as Tamandare Explorations Inc.)
(a development stage company)
CONSOLIDATED STATEMENTS OF OPERATIONS
| From June 7, 2007 | ||||||||||||
| (date of inception) | ||||||||||||
| Year ended December 31, | Through | |||||||||||
| 2011 | 2010 | December 31, 2011 | ||||||||||
| Costs and expenses: | ||||||||||||
| Research and development | $ | 1,158,167 | $ | 584,298 | $ | 1,951,954 | ||||||
| General and administrative | 2,220,361 | 1,344,390 | 4,255,247 | |||||||||
| 3,378,528 | 1,928,688 | 6,207,201 | ||||||||||
| Operating loss | (3,378,528 | ) | (1,928,688 | ) | (6,207,201 | ) | ||||||
| Gain on extinguishment of debt | - | - | 7,908 | |||||||||
| Interest expense, net | (91,585 | ) | (35,782 | ) | (195,387 | ) | ||||||
| Net loss | $ | (3,470,113 | ) | $ | (1,964,470 | ) | $ | (6,394,680 | ) | |||
| Net loss per common share, basic and diluted | $ | (0.16 | ) | $ | (0.18 | ) | ||||||
| Weighted average common shares outstanding, basic and diluted | 21,425,632 | 11,175,096 | ||||||||||
See notes to consolidated financial statements
| F-4 |
TONIX PHARMACEUTICALS HOLDING CORP.
(Formerly known as Tamandare Explorations Inc.)
(a development stage company)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' DEFICIENCY
| Deficit | ||||||||||||||||||||
| Accumulated | ||||||||||||||||||||
| Additional | During | |||||||||||||||||||
| Common stock | Paid in | Development | ||||||||||||||||||
| Shares | Amount | Capital | Stage | Total | ||||||||||||||||
| Shares issued to founders for intellectual property in June 2007 ($0.15 per share) | 589,014 | $ | 589 | $ | 87,161 | $ | - | $ | 87,750 | |||||||||||
| Shares issued to bankers for services in June 2007 ($0.15 per share) | 65,446 | 66 | 9,684 | - | 9,750 | |||||||||||||||
| Compensation related to restricted share awards issued to directors in November 2007 | - | - | 24,187 | - | 24,187 | |||||||||||||||
| Net loss | - | - | - | (537,001 | ) | (537,001 | ) | |||||||||||||
| Balance at December 31, 2007 | 654,460 | 655 | 121,032 | (537,001 | ) | (415,314 | ) | |||||||||||||
| Compensation related to cancelled restricted share awards in December 2008 | - | - | 72,563 | - | 72,563 | |||||||||||||||
| Net loss | - | - | - | (202,262 | ) | (202,262 | ) | |||||||||||||
| Balance at December 31, 2008 | 654,460 | 655 | 193,595 | (739,263 | ) | (545,013 | ) | |||||||||||||
| Conversion of senior convertible notes into capital stock in June 2009 ($0.03 per share) | 7,200,000 | 7,200 | 192,800 | - | 200,000 | |||||||||||||||
| Shares issued to directors in July 2009 ($0.15 per share) | 31,414 | 31 | 4,649 | - | 4,680 | |||||||||||||||
| Capital contribution in June 2009 | - | - | 23,725 | - | 23,725 | |||||||||||||||
| Net loss | - | - | - | (220,834 | ) | (220,834 | ) | |||||||||||||
| Balance at December 31, 2009 | 7,885,875 | 7,886 | 414,769 | (960,097 | ) | (537,442 | ) | |||||||||||||
| Conversion of demand notes into capital stock in July 2010 ($0.23 per share) | 2,094,547 | 2,095 | 477,905 | - | 480,000 | |||||||||||||||
| Conversion of accrued interest on demand notes into capital stock in July 2010 ($0.23 per share) | 301,430 | 301 | 68,777 | - | 69,078 | |||||||||||||||
| Issuance of capital stock in August to December 2010 ($0.23 per share) | 5,856,005 | 5,856 | 1,336,145 | - | 1,342,001 | |||||||||||||||
| Shares issued to founders for intellectual property in June 2010 ($0.23 per share) | 1,308,921 | 1,309 | 294,191 | - | 295,500 | |||||||||||||||
| Issuance of restricted shares to directors, employees and consultants in June to November 2010 ($0.24 per share) | 587,705 | 588 | 139,294 | - | 139,882 | |||||||||||||||
| Net loss | - | - | - | (1,964,470 | ) | (1,964,470 | ) | |||||||||||||
| Balance at December 31, 2010 | 18,034,483 | 18,035 | 2,731,081 | (2,924,567 | ) | (175,451 | ) | |||||||||||||
| Vesting and issuance of capital stock in January to September 2011 ($0.23 per share) | 2,670,548 | 2,670 | 609,330 | - | 612,000 | |||||||||||||||
| Vesting and issuance of restricted shares to directors, employees and consultants in February to April 2011 and vesting of restricted shares pursuant to Share Exchange in October 2011 | 1,961,636 | 1,962 | 433,689 | - | 435,651 | |||||||||||||||
| Common stock issued in connection with the share exchange transaction in October 2011 | 4,000,000 | 4,000 | (4,000 | ) | - | - | ||||||||||||||
| Common stock issued in October 2011 in exchange for services rendered ($0.36 per share) | 400,000 | 400 | 143,600 | - | 144,000 | |||||||||||||||
| Net loss | - | - | - | (3,470,113 | ) | (3,470,113 | ) | |||||||||||||
| Balance at December 31, 2011 | 27,066,667 | $ | 27,067 | $ | 3,913,700 | $ | (6,394,680 | ) | $ | (2,453,913 | ) | |||||||||
See notes to consolidated financial statements
| F-5 |
TONIX PHARMACEUTICALS HOLDING CORP.
(Formerly known as Tamandare Explorations Inc.)
(a development stage company)
CONSOLIDATED STATEMENTS OF CASH FLOWS
| From June 7, 2007 | ||||||||||||
| (date of inception) | ||||||||||||
| Year ended December 31, | Through | |||||||||||
| 2011 | 2010 | December 31, 2011 | ||||||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||||||
| Net loss | $ | (3,470,113 | ) | $ | (1,964,470 | ) | $ | (6,394,680 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||
| Depreciation | 9,300 | 3,854 | 17,312 | |||||||||
| Amortization of deferred financing costs | 53,377 | - | 53,377 | |||||||||
| Common stock issued in exchange for intellectual property | - | 295,500 | 383,250 | |||||||||
| Stock based compensation | 435,651 | 139,882 | 686,713 | |||||||||
| Gain on extinguishment of debt | - | - | (7,908 | ) | ||||||||
| Changes in operating assets and liabilities: | ||||||||||||
| Prepaid expenses | (79,117 | ) | (19,315 | ) | (102,430 | ) | ||||||
| Accounts payable | 377,453 | 294,132 | 695,198 | |||||||||
| Accrued interest | 38,306 | 32,691 | 38,306 | |||||||||
| Accrued expenses | (12,304 | ) | (34,789 | ) | 110,940 | |||||||
| Deferred rent payable | 9,909 | 19,174 | 29,083 | |||||||||
| Net cash used in operating activities | (2,637,538 | ) | (1,233,341 | ) | (4,490,839 | ) | ||||||
| CASH FLOWS FROM INVESTING ACTIVITIES: | ||||||||||||
| Purchase of furniture and equipment | (2,764 | ) | (34,279 | ) | (42,862 | ) | ||||||
| Repayment of security deposit | 3,156 | - | - | |||||||||
| Payment of restricted cash | (90 | ) | (60,087 | ) | (60,177 | ) | ||||||
| Net cash provided by (used in) investing activities | 302 | (94,366 | ) | (103,039 | ) | |||||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||||||
| Proceeds from demand notes | - | 50,000 | 480,000 | |||||||||
| Proceeds from notes payable | 500,000 | - | 700,000 | |||||||||
| Proceeds from Convertible Debentures | 1,501,000 | - | 1,501,000 | |||||||||
| Proceeds from the sale of capital stock | 612,000 | 1,342,001 | 1,954,001 | |||||||||
| Net cash provided by financing activities | 2,613,000 | 1,392,001 | 4,635,001 | |||||||||
| Net (decrease) increase in cash | (24,236 | ) | 64,294 | 41,123 | ||||||||
| Cash, beginning of the period | 65,359 | 1,065 | - | |||||||||
| Cash, end of period | $ | 41,123 | $ | 65,359 | $ | 41,123 | ||||||
| Non cash investing and financing activities: | ||||||||||||
| Senior convertible notes exchanged for preferred shares | $ | - | $ | 200,000 | $ | 200,000 | ||||||
| Capital contribution of accrued interest on convertible notes | $ | - | $ | - | $ | 23,725 | ||||||
| Demand notes together with accrued interest converted into capital stock | $ | - | $ | 549,078 | $ | 549,078 | ||||||
| Common stock issued for deferred financing costs | $ | 144,000 | $ | - | $ | 144,000 | ||||||
See notes to consolidated financial statements
| F-6 |
TONIX PHARMACEUTICALS HOLDING CORP.
(Formerly known as Tamandare Explorations Inc.)
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2011 AND 2010
NOTE 1 –BUSINESS AND RECAPITALIZATION
Tonix Pharmaceuticals Holding Corp. through its wholly owned subsidiary Tonix Pharmaceuticals, Inc. is attempting to develop safer and more effective versions of widely prescribed central nervous system ("CNS") drugs. While some new applications can use the commercially available form of the drug, in other cases reformulating the active ingredient improves its safety or effectiveness in treating the CNS condition. When formal development programs have proven successful in clinical tests, Tonix Pharmaceuticals, Inc. intends to seek marketing approval from the Food and Drug Administration ("FDA").
On August 16, 2010, Tonix Pharmaceuticals, Inc. formed Krele LLC ("Krele") in the state of Delaware. Krele is a limited liability corporation whose sole member is Tonix Pharmaceuticals Inc. Krele was established to commercialize products that are generic versions of predicate new drug application products or versions of drug efficacy study implementation products. The Company expects that its relationship to Krele will be similar to that of several other pharmaceutical companies and their subsidiaries that market generic versions of the parent's branded products at different periods in their product life-cycle.
On October 7, 2011, Tonix Pharmaceuticals, Inc. (formerly Krele Pharmaceuticals, Inc. incorporated on June 7, 2007 in the State of Delaware) and a publicly traded non-operating shell company Tamandare Explorations Inc. (“Tamandare”), incorporated under the laws of the State of Nevada, along with certain other parties executed and consummated a share exchange agreement (the “Share Exchange”). Pursuant to the Share Exchange, each share of Tonix Pharmaceuticals Inc.’s common stock was exchanged for 0.9 shares of Tamandare’s common stock and each share of Tonix Pharmaceuticals, Inc.’s Series A and B preferred stock was exchanged for 4.8 shares of Tamandare’s common stock. Upon completion of the Share Exchange, the Tonix Pharmaceuticals, Inc. shareholders, including holders of restricted shares, which were subject to accelerated vesting, received in exchange for all of their shares, an aggregate of 22,666,667 shares of Tamandare’s common stock and Tamandare’s existing stockholders retained 4,000,000 shares of common stock. The 22,666,667 shares issued to the Tonix Pharmaceuticals, Inc. shareholders constituted approximately 85% of Tamandare’s 26,666,667 issued and outstanding shares of common stock after the Share Exchange. Upon completion of the Share Exchange, Tonix Pharmaceuticals, Inc. became Tamandare’s wholly-owned subsidiary and in October 2011 Tamandare was renamed Tonix Pharmaceuticals Holding Corp. As the owners and management of Tonix Pharmaceuticals, Inc. obtained voting and operating control of Tamandare after the Share Exchange and Tamandare was non-operating, had no assets or liabilities and did not meet the definition of a business, the transaction has been accounted for as a recapitalization of Tonix Pharmaceuticals, Inc., accompanied by the issuance of its common stock for outstanding common stock of Tamandare, which was recorded at a nominal value. The accompanying financial statements and related notes give retroactive effect to the recapitalization as if it had occurred on June 7, 2007 (inception date) and accordingly all share and per share amounts have been adjusted.
NOTE 2 - SIGNIFICANT ACCOUNTING POLICIES
Basis of presentation:
The consolidated financial statements include the accounts of Tonix Pharmaceuticals Holding Corp. and its wholly owned subsidiaries, Tonix Pharmaceuticals, Inc. and Krele LLC (hereafter referred to as the “Company” or “Tonix”). All significant intercompany balances and transactions have been eliminated in consolidation.
As the Company is devoting substantially all of its efforts to establishing a new business, and while planned principal operations have commenced, there has been no revenue generated from sales, license fees or royalties, the Company is considered a development stage enterprise. Accordingly, the Company's consolidated financial statements are presented in accordance with authoritative accounting guidance related to a development stage enterprise. Financial position, results of operations and cash flows of a development stage enterprise are presented in conformity with generally accepted accounting principles that apply to established operating enterprises.
As a development stage enterprise, the Company's primary efforts are devoted to conducting research and development for the treatment of CNS diseases. The Company has experienced net losses and negative cash flows from operations since inception and expects these conditions to continue for the foreseeable future. In addition, the Company has both working capital and stockholders' deficiencies at December 31, 2011 and requires additional financing to fund future operations. Although, in the first quarter of 2012, the Company raised approximately $4,700,000 (see Note 11), it’s expected that cash used in operations will increase significantly over the next several years. The Company intends to raise additional capital to complete the development and commercialization of its current product candidates through equity or debt financing; however the Company does not have any commitments or definitive or binding arrangements for such funds. There can be no assurance that such funds, if available at all, can be obtained on terms reasonable to the Company. If the Company is unsuccessful in raising additional capital it will need to reduce costs and operations substantially. Further, the Company does not have any commercial products available for sale and has not generated revenues and there is no assurance that if approval of their products is received that the Company will be able to generate cash flow to fund operations. In addition, there can be no assurance that the Company's research and development will be successfully completed or that any product will be approved or commercially viable.
| F-7 |
TONIX PHARMACEUTICALS HOLDING CORP.
(Formerly known as Tamandare Explorations Inc.)
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2011 AND 2010
The above factors raise substantial doubt as to the Company's ability to continue as a going concern. The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern and do not include any adjustments that may result from the outcome of this uncertainty.
Use of estimates
The preparation of financial statements in accordance with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosures of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates. Significant estimates include the useful life of fixed assets and assumptions used in the fair value of stock-based compensation.
Research and Development costs
The Company outsources its research and development efforts and expenses related costs as incurred, including the cost of manufacturing product for testing, licensing fees and costs associated with planning and conducting clinical trials. The value ascribed to patents and other intellectual property acquired was expensed in 2007 and 2010 as research and development costs, as it related to particular research and development projects and had no alternative future uses.
Reclassifications
The accompanying 2010 financial statements together with cumulative amounts from inception have been reclassified to allocate professional services expenses to research and development and general and administrative expenses to be consistent with current year presentation.
Furniture and equipment
Furniture and equipment are stated at cost, less accumulated depreciation. Depreciation is calculated using the straight-line method over the asset's estimated useful life, which is three years for computer assets and five years for furniture and all other equipment. Expenditures for maintenance and repairs are expensed as incurred.
Income taxes
Deferred income tax assets and liabilities are determined based on the estimated future tax effects of net operating loss and credit carryforwards and temporary differences between the tax basis of assets and liabilities and their respective financial reporting amounts measured at the current enacted tax rates. The Company records an estimated valuation allowance on its deferred income tax assets if it is not more likely than not that these deferred income tax assets will be realized.
The Company recognizes a tax benefit from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by taxing authorities, based on the technical merits of the position. The tax benefits recognized in the consolidated financial statements from such a position are measured based on the largest benefit that has a greater than 50% likelihood of being realized upon ultimate settlement. As of December 31, 2011 and 2010, the Company has not recorded any unrecognized tax benefits.
| F-8 |
TONIX PHARMACEUTICALS HOLDING CORP.
(Formerly known as Tamandare Explorations Inc.)
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2011 AND 2010
Stock-based compensation
All stock-based payments to employees and to nonemployee directors for their services as directors, including grants of restricted stock and stock options, are measured at fair value on the grant date and recognized in the consolidated statements of operations as compensation or other expense over the relevant vesting period. Stock-based payments to nonemployees are recognized as an expense over the period of performance. Such payments are measured at fair value at the earlier of the date a performance commitment is reached or the date performance is completed. In addition, for awards that vest immediately and are non forfeitable the measurement date is the date the award is issued.
Per share data:
Basic and diluted net loss per common share is calculated by dividing net loss, by the weighted average number of outstanding shares of common stock, adjusted to give effect to the exchange ratio in the Share Exchange in October 2011 (see Note 1), which was accounted for as recapitalization of the Company.
In October 2011, upon completion of the share exchange referred to above, the Company issued Convertible Debentures in the amount of $2,075,000 which as of December 31, 2011 were convertible into approximately 3,985,000 common shares. In computing diluted net loss per share, no effect has been given to such shares as their effect would be antidilutive. See Notes 5 and 10 for subsequent issuance of securities.
NOTE 3 – FURNITURE AND EQUIPMENT
Furniture and equipment as of December 31, 2011 and 2010 is summarized as follows:
| 2011 | 2010 | |||||||
| Office furniture and equipment | $ | 42,862 | $ | 40,098 | ||||
| Less: accumulated depreciation | (17,312 | ) | (8,012 | ) | ||||
| $ | 25,550 | $ | 32,086 | |||||
Depreciation expense for the years ended December 31, 2011 and 2010 was $9,300 and $3,854, respectively.
NOTE 4 - RESTRICTED CASH
Restricted cash at December 31, 2011 and 2010 collateralizes a letter of credit in the amount of approximately $60,000 issued in connection with the lease of office space in New York City (see Note 6).
NOTE 5 - CONVERTIBLE DEBENTURES
On October 7, 2011, concurrently with the Share Exchange, the Company issued secured Convertible Debentures (“Convertible Debentures”) in the amount of $1,625,000 of which $1,125,000 were sold to certain investors for aggregate cash proceeds of $1,065,000, net of selling commissions to a placement agent of $40,000 and $20,000 of legal fees, and $500,000 were exchanged for 8% Notes Payable (“Notes Payable”) issued on September 9, 2011. In addition, 400,000 shares of common stock with the fair market value of $144,000 were issued to a second placement agent. On November 16, the Company issued Convertible Debentures in the amount of $450,000 for aggregate cash proceeds of $436,000, net of selling commissions to a third placement agent of $14,000.
The Convertible Debentures mature on the earlier of (i) one year from the date of issuance or (ii) the date of closing of a private placement of equity, equity equivalent, convertible debt or debt financing in which the Company receives gross proceeds, in one or more transactions, of at least $3,425,000 (a “Subsequent Financing”). The Convertible Debentures bear interest at 8% per annum and are convertible at the holder’s option into a Subsequent Financing. In the event that a Subsequent Financing has not occurred within 12 months from the date of issuance of the Convertible Debentures, the holder has the option to convert into a number of shares of the Company's common stock equal to 1% of the Company's shares of common stock on a fully diluted basis for every $125,000 of Convertible Debentures (the “Conversion Shares”) or an aggregate of approximately 3,985,000 shares based on the outstanding shares of the Company common stock as of December 31, 2011. A Subsequent Financing comprised of Units consisting of common stock and warrants took place on January 20, 2012 and $1,925,000 of debentures were exchanged for Units (see Note 10). The remaining $150,000 of debentures were repaid. As a result of the exchange, $1,925,000 principal amount of debentures are classified as a non-current liability in the accompanying balance sheet at December 31, 2011.
| F-9 |
TONIX PHARMACEUTICALS HOLDING CORP.
(Formerly known as Tamandare Explorations Inc.)
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2011 AND 2010
Upon conversion or repayment of the Convertible Debenture, the holder is entitled to receive, at the holder’s option, either (i) a warrant (the “Warrant”), which has a three year term and is exercisable at the offering price in a Subsequent Financing, to purchase such number of shares of the Company's common stock equal to the principal amount of the Convertible Debenture divided by the offering price in a Subsequent Financing, (the “Warrant Shares”) or (ii) shares of the Company's common stock equal to 33% of the principal amount of the Debenture divided by the offering price in a Subsequent Financing (the “Incentive Shares”). The value of the Warrant Shares or Incentive Shares will be measured and number of such shares determined upon occurrence of any Subsequent Financing. The Conversion Shares, Warrant Shares and Incentive Shares are entitled to piggyback registration rights. Upon the Subsequent Financing on January 20, 2012, the holders $1,925,000 principal amount of Convertible Debentures elected to receive 240,000 Warrants exercisable at $1 per share and 536,250 Incentive Shares, and holders of the remaining $150,000 principal amount of Convertible Debentures, which were redeemed, received 25,000 Warrants exercisable at $1 per share and 57,750 Incentive Shares. The value of the Warrants and Incentive Shares will be charged to operations in the first quarter of 2012.
In addition to selling commissions paid to the placement agents on the sale of certain Convertible Debentures, the placement agents received warrants which expire in October 2013 and November 2013, respectively, and are exercisable at the offering price in a Subsequent Financing to purchase shares of the Company's common stock equal to 3% and 9%, respectively, of the gross proceeds delivered by purchasers introduced by such placement agents divided by the purchase price per share in the Subsequent Financing. In the event that the Subsequent Financing has not occurred within 12 months from the date of issuance of the Convertible Debentures, the placement agents will receive, in lieu of the warrants, shares of common stock equal to 3% and 9%, respectively, of the number of shares of the Company's common stock such purchasers are entitled to receive upon conversion of their Convertible Debentures or an aggregate of approximately 88,000 shares based on the outstanding shares of the Company's common stock as of December 31, 2011. The Company recognized a liability to placement agents to issue shares of its common stock based on their fair value of approximately $32,000 as of December 31, 2011. Upon the Subsequent Financing on January 20, 2012, the placement agents become entitled to receive 30,750 warrants exercisable at $1.00 per share.
The following expenses in connection with the issuance of Convertible Debenture are recorded as deferred financing costs: fair value of 400,000 shares of the Company’s common stock issued to the placement agent valued at $144,000, cash payments to the placement agents of $54,000, legal expenses of $20,000 and fair value of the liability to placement agent to issue the Company’s shares of common stock in the amount of $32,000. The deferred financing costs are being amortized using the effective interest method over the twelve month term of the Convertible Debentures. During the year ended December 31, 2011, amortization of deferred financing costs amounted to approximately $53,000 and charged to interest expenses in the statement of operations. The unamortized balance will be charged to operations in connection with the extinguishment of the debentures resulting from their exchange for the Units and repayment in 2012.
Pursuant to a Pledge and Security Agreement and Subsidiary Guaranty, the Company granted the Debenture holders a first priority lien on all its assets.
NOTE 6 - COMMITMENTS
Operating leases
On September 28, 2010, the Company entered into a five-year lease for office space in New York City, with monthly payments escalating from approximately $10,000 in first year to approximately $11,000 in fifth year. The Company received a rent credit of $9,420 in each of the months of November 2010, December 2010 and January 2011. The Company has posted a letter of credit in the amount of approximately $60,000 for the benefit of the landlord which is collateralized by a money market account (see Note 7 - Restricted Cash).
| F-10 |
TONIX PHARMACEUTICALS HOLDING CORP.
(Formerly known as Tamandare Explorations Inc.)
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2011 AND 2010
Future minimum lease payments under the operating lease are as follows:
| Year Ending December 31, | |||||
| 2012 | $ | 124,370 | |||
| 2013 | 127,889 | ||||
| 2014 | 131,513 | ||||
| 2015 | 100,719 | ||||
| $ | 484,491 | ||||
Rent expense charged to operations, which differs from rent paid due to the rent credits and to increasing amounts of base rent, is calculated by allocating total rental payments on a straight-line basis over the term of the lease. During the years ended December 31, 2011 and 2010, rent expense was $128,228 and $42,570, respectively and as of December 31, 2011 and 2010 deferred rent payable was $29,083 and $19,174, respectively. The Company utilized office space in New York City provided by founders without remuneration until October 2010.
Consulting agreements
In June 2010, the Company entered into a two-year consulting agreement with L&L Technologies, an entity controlled by a member of the Company’s Board of Directors, for scientific and medical consulting services. In consideration for such services, L&L Technologies will receive $96,000 per annum and 1,026,194 shares of restricted common stock which were granted at the inception of the agreement. The consulting agreement renews automatically for subsequent terms of one year at $96,000 per annum. The restricted shares vest as follows: 25% on the grant date (June 4, 2010) and 25% on each of the first and second annual anniversaries of the grant date and, if the consulting agreement is renewed, 25% on the third anniversary of the grant date. Vesting of the unvested 513,097 restricted shares accelerated on October 7, 2011, the date of the Share Exchange.
In June 2010, the Company entered into a two-year consulting agreement with Lederman & Co., an entity controlled by a member of the Company’s Board of Directors, for clinical development, strategic, management and operational consulting services. In consideration for such services, Lederman & Co. will receive $250,000 per annum and 261,784 shares of restricted common stock which were granted at the inception of the agreement. The consulting agreement renews automatically for subsequent terms of one year at $250,000 per annum. The restricted shares vest as follows: 20% on the grant date (June 4, 2010) and 20% on each of the first and second anniversaries of the grant date and, if the consulting agreement is renewed, 20% on each of the third and fourth anniversaries of the grant date. Vesting of the unvested 157,087 restricted shares accelerated on October 7, 2011, the date of the Share Exchange.
In June 2010, the Company entered into an agreement with Burns McClellan, Inc. to provide media and investor relations services, including preparation of investor presentations and press releases, media outreach and training and investor targeting and introductions, for a fee of $20,000 per month, plus expenses. The agreement was terminated in January 2011.
In October 2010, the Company entered into an agreement with Frost & Sullivan to prepare an assessment of the U.S. fibromyalgia market, including current market size and historical and projected growth rates, as well as a formal presentation supporting their findings for a fee of $109,400, all of which was recognized in 2010.
In July 2011, the Company entered into an agreement with Catalent Pharma Solutions, LLC to investigate, for $58,080, the feasibility of developing the active pharmaceutical ingredient (“API”) used in TNX-102, one of the Company’s product candidates, for use in a new, proprietary formulation
In August 2011, the Company entered into an agreement with Porter, LeVay & Rose, Inc. to provide media and investor relations services, including preparation of investor presentations and press releases, media outreach and training and investor targeting and introductions, for a fee of $12,000 per month, plus expenses. Also in August 2011, the Company entered into an agreement with JFC Technologies, LLC (“JFC”) for product development work for an initial fee of $75,000, of which $35,000 was paid upon signing. In September 2011, JFC was acquired by Cyalume Specialty Products, Inc. (“Cyalume”) and the Company’s agreement was transferred to Cyalume. Additionally, in August 2011 the Company authorized the initiation of stage 2 work pursuant to a contract with Lipocine Inc. with respect to a research and development project for reformulation work on TNX-102 for a fee of $235,000, which work started in the third quarter of 2011.
| F-11 |
TONIX PHARMACEUTICALS HOLDING CORP.
(Formerly known as Tamandare Explorations Inc.)
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2011 AND 2010
In September 2011, the Company entered into two contracts with Pharmanet Canada for contract research work with respect to the development of methods to measure the active ingredient of TNX-102 in blood and urine. The full cost of the work to be performed is approximately $90,000. Payment is due in three installments based on the achievement of certain performance milestones. Also, in September 2011, the Company entered into a contract with Pharmanet Canada for contract research work with respect to the pharmacokinetic study for TNX-102. The full cost of the work to be performed is $637,231. Payment is due in four installments based on the achievement of certain performance milestones.
In October 2011, the Company entered into an agreement with Applied Pharma Research to develop, and perform an exploratory pharmacokinetic study on a new formulation of the API used in TNX-102 for an approximate cost of $180,000.
Employment agreements
In 2010, the Company entered into employment agreements with the Chief Operating Officer and the Vice President of Marketing which expire in August 2012 and June 2012, respectively. Under the terms of the employment agreements, the Chief Operating Officer and the Vice President of Marketing shall receive annual base compensation of $250,000 and $150,000, respectively, which shall be adjusted to $320,000 and $200,000, respectively, or such other rate as the Board may designate from time to time, upon completion of an initial public offering with net proceeds of at least $15,000,000. The agreements will be automatically renewed for additional one-year periods (the "Renewal Terms") unless either party notifies the other in writing of its intention not to renew within 90 days prior to the expiration of the Initial Term or any Renewal Terms. Upon termination without cause, as defined in the agreements, the executives will continue to receive compensation for up to nine months if termination is in connection with or following an initial public offering.
In February 2011, the Company entered into an employment agreement with the Chief Business Officer which expires in February 2013. Under the terms of the employment agreement, the Chief Business Officer shall receive annual base compensation of $150,000 which shall increase, with a retroactive adjustment, upon the completion of an underwritten public offering, as defined, or certain other events. The employment agreement will be automatically renewed for additional renewal terms unless either party notifies the other in writing of its intention not to renew within 90 days prior to the expiration of the initial term or any renewal terms. Upon termination without cause, as defined in the employment agreement, the Chief Business Officer will continue to receive compensation for six months, or nine months if termination is in connection with or following certain events.
In April 2011, the Company terminated existing employment agreements with the three executive employees referred to in the first two paragraphs above and entered into new employment agreements which stipulate such employees will receive minimum wage salary ($7.25 per hour) for a 40 hour week until the Company receives new capital of at least $500,000 through the sale of equity securities. The expiration dates of the new agreements remain the same as the terminated agreements. In addition, the Chief Business Officer assumed the title of Chief Operating Officer and the Chief Operating Officer assumed the title of Chief Financial Officer and Chief Administrative Officer and the Vice President of Marketing assumed the title of Vice President of Strategy. Upon receipt of $500,000 or more in new capital, the employees will receive a lump sum payment in the amount of $50,000 each for the Chief Operating Officer and Chief Financial Officer and $30,000 for the Vice President of Strategy. Further, base salaries for all three employees will be increased with a retroactive adjustment upon the completion of an underwritten offering, as defined, or certain other events. All other terms remain the same. In October 2011, the position of Vice President of Strategy was eliminated and the employment agreement was terminated. In conjunction with this event, the Company paid $37,500 in December 2011 in exchange for the release from future obligations. In February 2012, the Company terminated its employment agreement with its Chief Financial Officer and in accordance with the agreement paid such officer approximately $88,000.
In July 2011, the Company entered into agreements with the executive employees to defer payment of the lump sum amounts referred to above until the closing of a private placement of securities, as defined. In addition, salaries of the Chief Financial Officer and Chief Operating Officer were adjusted to $175,000 per annum effective August 2011. The salary of the Chief Operating Officer shall increase to $250,000 per annum on the first anniversary of the Share Exchange provided that the Company has raised at least $500,000 in additional equity securities.
| F-12 |
TONIX PHARMACEUTICALS HOLDING CORP.
(Formerly known as Tamandare Explorations Inc.)
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2011 AND 2010
NOTE 7 - INCOME TAXES
There is no provision for federal or state income taxes for the years ended December 31, 2011 and 2010 since the Company has established a valuation allowance equal to the total deferred tax asset related to losses incurred during such periods.
Deferred tax assets and liabilities and related valuation allowance as of December 31, 2011 and 2010 are as follows:
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| Deferred tax assets: | ||||||||
| Organization costs | $ | 733 | $ | 2,494 | ||||
| Research and development credit carryforward | 6,188 | 6,188 | ||||||
| Net operating loss carryforwards | 2,329,829 | 1,107,688 | ||||||
| Other | 132,482 | 121,091 | ||||||
| Total deferred tax assets | 2,469,232 | 1,237,461 | ||||||
| Deferred tax liabilities: | ||||||||
| Restricted stock compensation(1) | - | (148,871 | ) | |||||
| Net deferred tax assets | 2,469,232 | 1,088,590 | ||||||
| Valuation allowance | (2,469,232 | ) | (1,088,590 | ) | ||||
| Net deferred tax assets | $ | 0 | $ | 0 | ||||
| (1) | Relates to restricted stock grants for which Internal Revenue Code ("IRC") Section 83(b) elections were filed in 2010, resulting in tax deductions in excess of related compensation expense for financial reporting purposes in 2010. |
Based on the Company's historical losses and its expectation of continuation of losses for the foreseeable future, the Company has determined that it is not more likely than not that the deferred tax assets will be realized and, accordingly, has provided a valuation allowance. The increase in the valuation allowance for the years ended December 31, 2011 and 2010 was $1,380,642 and $783,696, respectively.
At December 31, 2011, the Company has available unused net operating loss carryforwards of approximately $5.8 million that expire from 2027 to 2031 for federal tax purposes and the same amount for New Jersey state tax purposes, which expire from 2014 to 2018. The Company also has approximately $3 million of net operating loss carryforwards for New York state purposes expiring in 2031. These net operating loss carryforwards may be subject to annual limitations in their use in accordance with IRC Section 382. Accordingly, the extent to which the net operating loss carryforwards can be used to offset future taxable income may be limited. At December 31, 2011, the Company has a research and development credit carryforward of $6,188 for federal tax purposes that expires in 2027.
The Company's federal and state tax returns remain open and subject to examination by the tax authorities for the tax years 2008 and 2007, respectively through 2011.
A reconciliation of the effect of applying the federal statutory rate and the effective income tax rate used to calculate the Company's income tax provision is as follows:
| F-13 |
TONIX PHARMACEUTICALS HOLDING CORP.
(Formerly known as Tamandare Explorations Inc.)
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2011 AND 2010
| Year Ended December 31, | ||||||||
| 2011 | 2010 | |||||||
| Statutory federal income tax | (34.0 | )% | (34.0 | )% | ||||
| State income tax, net of federal tax effect | (5.9 | )% | (5.9 | )% | ||||
| Permanent difference | 0.0 | % | 5.0 | % | ||||
| Increase in valuation allowance | 39.9 | % | 34.9 | % | ||||
| Income tax provision | 0 | % | 0 | % | ||||
NOTE 8 – STOCK PLAN
In June and August 2010, respectively, the board of directors and stockholders of Tonix Pharmaceuticals, Inc. approved, and in December 2010 and February 2011, the board of directors amended, the terms and provisions of the 2010 Stock Plan ("Plan") whereby the Company reserved 4,564,641 shares of its Common Stock for issuance pursuant to the Plan. The Plan allowed for grants of options to purchase shares of Common Stock and awards of restricted Common Stock to employees, officers, directors, consultants and advisors of the Company.
In 2010, the Company granted shares of restricted Common Stock under the Plan to employees ("Employee Granted Shares") as follows: 196,359 shares to the Chief Operating Officer, 109,088 shares to the Vice President of Clinical Development, 130,906 shares to the Vice President of Marketing and 196,359 shares to the Chief Medical Officer. Employee Granted Shares vest: 20% on the grant date and 20% on each of the first, second, third and fourth anniversaries of the grant date. Upon termination of the Chief Medical Officer's employment with Tonix, 157,087 unvested shares held by him were forfeited and he retained 39,272 shares of fully vested Common Stock. Upon termination of the Vice President of Clinical Development's employment with Tonix, 87,270 unvested shares held by him were forfeited and he retained 21,818 shares of fully vested Common Stock.
In 2010, the Company granted 1,288,112 shares of restricted Common Stock under the Plan to consultants.
In 2010, the Company granted 556,786 shares of restricted Common Stock under the Plan to directors and also granted 52,362 shares of restricted Common Stock under the Plan to members of the Scientific Advisory Board which vest: 25% on the grant date and 25% on each of the first, second and third anniversaries of the grant date.
In February 2011, the Company granted shares of restricted Common Stock to employees as follows: 196,359 shares to the Chief Business Officer and 130,906 shares to the incoming President of Krele. The shares vest: 20% on the grant date and 20% on each of the first, second, third and fourth anniversaries of the grant date. In August 2011, upon resignation of the President of Krele, 104,725 unvested shares were forfeited.
In March and April 2011, the Company granted 19,636 and 21,818 shares of restricted Common Stock, respectively, to newly appointed members of the Scientific Advisory Board and the Board of Directors which vest: 25% on the grant date and 25% on each of the first, second and third anniversaries of the grant date.
Following is a summary of the status of the Company's nonvested restricted stock as of December 31, 2011 and the changes during the years 2010 and 2011:
| F-14 |
TONIX PHARMACEUTICALS HOLDING CORP.
(Formerly known as Tamandare Explorations Inc.)
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2011 AND 2010
| Weighted | ||||||||
| Number of | Average | |||||||
| Restricted | Grant-Date | |||||||
| Nonvested Restricted Stock | Shares | Fair Value | ||||||
| Nonvested at January 1, 2010 | ||||||||
| Granted | 2,529,971 | $ | 0.23 | |||||
| Vested | (587,767 | ) | $ | 0.23 | ||||
| Forfeited | (244,357 | ) | $ | 0.23 | ||||
| Nonvested at December 31, 2010 | 1,697,847 | $ | 0.23 | |||||
| Granted | 368,718 | $ | 0.23 | |||||
| Vested prior to Share Exchange | (564,858 | ) | $ | 0.23 | ||||
| Vested pursuant to Share Exchange | (1,396,982 | ) | $ | 0.23 | ||||
| Forfeited | (104,725 | ) | $ | 0.23 | ||||
| Nonvested at December 31, 2011 | 0 | $ | 0 | |||||
Restricted stock is not considered to be issued until the stock vests.
The Company recognized share-based compensation expense of $139,063 prior to Share Exchange and remaining expense of $296,588 was recognized on October 7, 2011, the date of Share Exchange, upon which all nonvested restricted shares vested and the Plan ceased to exist. Stock based compensation expense for the year ended December 31, 2010 was $139,882.
NOTE 9 - RELATED PARTY TRANSACTIONS
Dr. Seth Lederman and Dr. Donald Landry are primary founders of the Company and serve on the board of directors. They have entered into various transactions with the Company through several companies under their control, including L&L Technologies and Lederman & Co as described in Note 6.
During 2007, the Company issued senior convertible promissory notes (the "Senior Convertible Notes") pursuant to the Note Purchase Agreements among the Company and National Holdings Corporation, Lederman & Co., LLC, Eli Lederman PhD and Dr. Seth Lederman, all but one of whom are direct or indirect stockholders of the Company (collectively referred to herein as the "Noteholders"), in the amount of $50,000 per Senior Convertible Note, or $200,000 in the aggregate. The Senior Convertible Notes bore interest at the rate of 8% per annum and were payable together with the interest accrued thereon on the two year anniversary of the Senior Convertible Notes. The outstanding principal and interest accrued thereon were to be automatically converted into fully paid shares of capital stock upon the closing of a Qualified Financing of capital stock or securities convertible into preferred stock which resulted in gross proceeds of at least $2,000,000.
In June 2009, although a Qualified Financing had not occurred, the Noteholders agreed to exchange the Senior Convertible Notes for shares of capital stock of the Company at the rate of one share of capital per $0.13 of the outstanding principal balance of such notes. The accrued interest on the notes in the amount of $31,633 was forgiven. The excess of the carrying value of the notes including accrued interest over the fair value of the capital stock for which they were exchanged amounted to $31,633 of which $23,725, representing the excess related to the Noteholders who are direct or indirect stockholders, has been accounted for as a capital contribution and credited to additional paid-in capital and the remaining $7,908 was recorded as a gain on extinguishment of debt. Interest expense relating to the Senior Convertible Notes for the year ended December 31, 2009 was $8,044.
In 2007, Lederman & Co. loaned the Company $10,000. On December 19, 2008, the Company issued to Lederman & Co. a demand note in the amount of $280,000, which included new cash proceeds of $270,000 as well as the amount loaned in 2007, with interest accruing on the total demand note balance commencing December 19, 2008. On December 7, 2009, the Company borrowed an additional $150,000 from Lederman & Co. and issued a demand note. The principal balance of the demand notes outstanding as of December 31, 2009 was $430,000 with accrued interest owed at December 31, 2009 of $36,387. On March 5, 2010, the Company issued to Dr. Donald Landry a demand note in the amount of $50,000. The demand notes accrue interest at the rate of 12% per annum.
On July 30, 2010, the demand notes and all interest accrued thereon were converted into shares of capital stock. Demand notes held by Lederman & Co. totaling $430,000 and accrued interest thereon of $66,629 were converted into 2,166,444 shares of capital stock, at a conversion price of $0.23 per share of capital stock. The demand note held by Donald Landry totaling $50,000 and accrued interest thereon of $2,449 was converted into 228,835 shares of capital stock, at a conversion price of $0.23 per share of capital stock.
| F-15 |
TONIX PHARMACEUTICALS HOLDING CORP.
(Formerly known as Tamandare Explorations Inc.)
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2011 AND 2010
On September 9, 2011, the Company sold $500,000 principal amount of 8% convertible notes (the “Notes”) to members of the board of directors and their related parties. The Notes were due one year from the date of issuance, and were exchangeable for a future financing (the “New Financing”) at the option of the holders. Interest is payable on either the maturity date or on the date the Notes are exchanged into the New Financing, or such accrued interest can be converted into the New Financing. On October 7, 2011, the Notes were exchanged into debentures issued by the Company concurrently with the Share Exchange (see Note 5).
Interest expense on the demand notes for the years ended December 31, 2010 and 2009 was $32,691 and $35,267, respectively.
NOTE 10 - SUBSEQUENT EVENTS
The Company amended and restated its Bylaws and Articles of Incorporation on February 16, 2012 and among other changes, increased the number of authorized shares of common stock, $0.001 par value to 150,000,000. Additionally, the Company is now authorized to issue 5,000,000 shares of preferred stock, $0.001 par value with such designations, references and participating, optional or other special rights and qualifications, limitations or restrictions thereof as shall be determined by the Company’s Board of Directors.
On February 12, 2012 the Company’s Board of Directors approved the 2012 Incentive Stock Options Plan (the “Plan”). The Plan provides for the issuance of options to purchase up to 4,000,000 shares of the Company’s common stock to officers, directors, employees and consultants of the Company. Under the terms of the Plan the Company may issue Incentive Stock Options as defined by the Internal Revenue Code to employees of the Company only and nonstatutory options. The Board of Directors of the Company determines the exercise price, vesting and expiration period of the grants under the Plan. However, the exercise price of an Incentive Stock Option should not be less than 110% of fair value of the common stock at the date of the grant for a 10% or more stockholder and 100% of fair value for a grantee who is not 10% stockholder. The fair value of the common stock is determined based on quoted market price or in absence of such quoted market price, by the Board of Directors in good faith. Additionally, the vesting period of the grants under the Plan should not be more than five years and expiration period not more than ten years. The Company reserved 4,000,000 shares of its common stock for future issuance under the terms of the Plan.
Subsequent financing
On January 20, 2012, the Company issued an aggregate of 172.118 units (“Units”) to certain investors (the “Purchasers”) for aggregate cash proceeds of $2,377,950 and $1,925,000 in previously issued Convertible Debentures of the Company that were exchanged for Units (the “Financing”). On March 1, 2012, the Company issued an aggregate of 92.5926 units to certain investors for aggregate cash proceeds of $2,314,815.
Each Unit had a purchase price of $25,000 per Unit and consisted of twenty five thousand (25,000) shares of the Company’s common stock, a Class A Warrant to purchase twenty five thousand (25,000) shares of Common Stock (the “Class A Warrants”), and a Class B Warrant to purchase up to twenty five thousand (25,000) shares of Common Stock (the “Class B Warrants” and together with the Class A Warrants, the “Warrants”).
The Class A Warrants have an exercise price of $1.25 per share of common stock and will be exercisable for a period of five years from the date of issuance. The Class B Warrants are exercisable automatically on their expiration date by cashless exercise or expire without exercise. In the event that the average of the Company’s daily volume weighted average price is below $0.75 during the 10 trading days after the Announcement Date (as hereinafter defined) (the “Measuring Period”), then the holder will be entitled to receive additional shares of the Company’s Common Stock upon the exercise of the Class B Warrants on the expiration date, which is the 12th trading day after the Announcement Date. In the event that the Company’s average daily volume weighted average price is at or above $0.75 during the Measuring Period, the Class B Warrants will expire unexercised. The Announcement Date is the earlier of (1) the date on which the Company announces via press release the results of the pharmacokinetic study of its TNX-102 drug formulation; or (2) June 1, 2012.
In connection with the Financing, the Company paid a placement agent (the “Agent”) an aggregate cash payment of $466,777, which represented an 8% commission and a 2% non-accountable expense allowance of the gross proceeds delivered by Purchasers in the Financing. In addition, the Agent earned warrants to purchase shares of Common Stock equal to 10% of the gross proceeds delivered by Purchasers in the Financing (the “Agent Warrants”), which have an exercise price of $1.25 per share of common stock, exercisable for a period of seven years, contain customary anti-dilution protection and are entitled to piggy-back registration rights.
| F-16 |
TONIX PHARMACEUTICALS HOLDING CORP.
(Formerly known as Tamandare Explorations Inc.)
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2011 AND 2010
Pursuant to the Warrants, no Purchaser may exercise such Purchaser’s Warrant if such exercise would result in the Purchaser beneficially owning in excess of 4.99% of the Company’s then issued and outstanding common stock. A Purchaser may, however, increase or decrease this limitation (but in no event exceed 9.99% of the number of shares of Common Stock issued and outstanding) by providing the Company with 61 days’ notice that such holder wishes to increase or decrease this limitation.
In connection with the Financing, the Company entered into a Registration Rights Agreements with Purchasers. The Company is required to file a registration statement registering for resale the common stock included in the Units and the common stock underlying the Warrants and the Agent Warrants to be filed no later than 60 days from the date of termination of the Financing on March 1, 2012 and must be declared effective no later than 120 days from the date of termination of the Financing (June 29, 2012). The Company is required to maintain the effectiveness of the registration statement from its effective date unless all securities registered under the registration statement have been sold or are otherwise able to be sold. If the Company fails to comply with the registration statement filing or effective date requirements, the Company is required to pay the investors a fee equal to 1.0% of the Purchaser’s investment, for each 30-day period of delay, subject to a maximum payment of 10% to each Purchaser.
The Company determined the offering price for the purpose of calculation of number of Warrants or Incentive Share to be issued to Convertible Debenture holders and warrants to be issued the placement agents of Convertible Debentures to be $1.00 (see Note 5).
| F-17 |
ITEM 9 - CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURES.
On October 7, 2011, we dismissed MaloneBailey LLP (“MaloneBailey”), as our independent registered public accounting firm. The reports of MaloneBailey on our financial statements for each of the past two fiscal years contained no adverse opinion or a disclaimer of opinion and were not qualified or modified as to uncertainty, audit scope or accounting principles, except as that the reports of MaloneBailey for the fiscal years ended December 31, 2010 and 2009 indicated conditions which raised substantial doubt about the Company’s ability to continue as a going concern. The decision to change independent accountants was approved by our Board of Directors on October 7, 2011.
During our two most recent fiscal years and through the date of this report, we have had no disagreements with MaloneBailey on any matter of accounting principles or practices, financial statement disclosure, or auditing scope or procedure, which disagreements, if not resolved to the satisfaction of MaloneBailey, would have caused it to make reference to the subject matter of such disagreements in its report on our financial statements for such periods.
During our two most recent fiscal years and through the date of termination of MaloneBailey, there have been no reportable events as defined under Item 304(a)(1)(v) of Regulation S-K adopted by the SEC.
Our Board of Directors appointed EisnerAmper LLP (“EisnerAmper”) as our new independent registered public accounting firm effective as of October 7, 2011. During the two most recent fiscal years and through the date of our engagement, we did not consult with EisnerAmper regarding either (1) the application of accounting principles to a specified transaction, either completed or proposed, or the type of audit opinion that might be rendered on our financial statements, or (2) any matter that was either the subject of a disagreement (as defined in Regulation S-K Item 304(a)(1)(v)), during the two most recent fiscal years.
Prior to engaging EisnerAmper, EisnerAmper did not provide our company with either written or oral advice that was an important factor considered by our company in reaching a decision to change our independent registered public accounting firm from MaloneBailey to EisnerAmper.
ITEM 9A – CONTROLS AND PROCEDURES
(a) Evaluation of disclosure controls and procedures.
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures pursuant to Rule 13a-15 under the Exchange Act. In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives. In addition, the design of disclosure controls and procedures must reflect the fact that there are resource constraints and that management is required to apply its judgment in evaluating the benefits of possible controls and procedures relative to their costs.
Based on management’s evaluation, our Chief Executive Officer and Chief Financial Officer concluded that, as a result of the material weaknesses described below, as of December 31, 2011, our disclosure controls and procedures are not designed at a reasonable assurance level and are ineffective to provide reasonable assurance that information we are required to disclose in reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in SEC rules and forms, and that such information is accumulated and communicated to our management, including our chief executive officer and chief financial officer, as appropriate, to allow timely decisions regarding required disclosure. The material weaknesses, which relate to internal control over financial reporting, that were identified are:
| a) | We did not have sufficient personnel in our accounting and financial reporting functions. As a result we were not able to achieve adequate segregation of duties and were not able to provide for adequate reviewing of the financial statements. This control deficiency, which is pervasive in nature, results in a reasonable possibility that material misstatements of the financial statements will not be prevented or detected on a timely basis; and |
| b) | We did not maintain sufficient personnel with an appropriate level of technical accounting knowledge, experience, and training in the application of U.S. GAAP commensurate with our complexity and our financial accounting and reporting requirements. This control deficiency is pervasive in nature. Further, there is a reasonable possibility that material misstatements of the financial statements including disclosures will not be prevented or detected on a timely basis as a result. |
We are committed to improving our accounting and financial reporting functions. As part of this commitment, we will create a segregation of duties consistent with control objectives and will look to hire additional personnel with technical accounting expertise, including a Chief Financial Officer and Controller, in the second quarter of 2012, to appropriately address non-routine or complex accounting matters. In addition, we have engaged an outside accounting consultant to provide additional knowledgeable personnel with technical accounting expertise to further support the current accounting personnel at the Company.
Management believes that hiring additional knowledgeable personnel with technical accounting expertise will remedy the following material weaknesses: (A) lack of sufficient personnel in our accounting and financial reporting functions to achieve adequate segregation of duties; and (B) insufficient personnel with an appropriate level of technical accounting knowledge, experience, and training in the application of U.S. GAAP commensurate with our complexity and our financial accounting and reporting requirements.
Management believes that the hiring of additional personnel who have the technical expertise and knowledge with the non-routine or technical issues we have encountered in the past will result in both proper recording of these transactions and a much more knowledgeable finance department as a whole. Due to the fact that our accounting staff consists of an interim Chief Financial Officer and a bookkeeper, additional personnel will also ensure the proper segregation of duties and provide more checks and balances within the department. Additional personnel will also provide the cross training needed to support us if personnel turnover issues within the department occur. We believe this will eliminate or greatly decrease any control and procedure issues we may encounter in the future.
We will continue to monitor and evaluate the effectiveness of our disclosure controls and procedures and our internal controls over financial reporting on an ongoing basis and are committed to taking further action and implementing additional enhancements or improvements, as necessary and as funds allow.
(b) Changes in internal control over financial reporting.
We regularly review our system of internal control over financial reporting and make changes to our processes and systems to improve controls and increase efficiency, while ensuring that we maintain an effective internal control environment. Changes may include such activities as implementing new, more efficient systems, consolidating activities, and migrating processes. Effective October 7, 2011, we entered into a reverse merger transaction, pursuant to which our sole officer and director resigned, and new officers and directors were appointed. Other than the change in the officers and the board of directors, there were no changes in our internal control over financial reporting that occurred during the quarter ended December 31, 2011 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| 46 |
(c) Management’s report on internal control over financial reporting.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Exchange Act Rule 13a-15(f). Management conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on this evaluation, management concluded that our internal control over financial reporting was ineffective as of December 31, 2011 for the reasons discussed above.
This annual report does not include an attestation report by EisnerAmper LLP, our independent registered public accounting firm regarding internal control over financial reporting. As a smaller reporting company, our management's report was not subject to attestation by our registered public accounting firm pursuant to rules of the Securities and Exchange Commission that permit us to provide only management's report in this annual report.
ITEM 9B – OTHER INFORMATION
None.
| 47 |
PART III
ITEM 10 – DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The names of our executive officers and directors and their age, title, and biography as of March 20, 2012 are set forth below:
| Name | Age | Title | ||
| Seth Lederman | 54 | President, CEO and Chairman of the Board of Directors | ||
| Benjamin Selzer | 34 | Interim Chief Financial Officer, Chief Operating Officer, Secretary and Treasurer | ||
| Stuart Davidson | 54 | Director | ||
| Patrick Grace | 55 | Director | ||
| Donald Landry | 57 | Director | ||
| Ernest Mario | 73 | Director | ||
| Charles Mather IV | 51 | Director | ||
| John Rhodes | 55 | Director |
Directors are elected annually and hold office until the next annual meeting of the stockholders of the Company and until their successors are elected. Officers are elected annually and serve at the discretion of the Board of Directors.
Seth Lederman, MD became our President, Chief Executive Officer, Chairman of the Board and a Director in October 2011. Dr. Lederman founded Tonix Sub in June of 2007 and has acted as its Chairman of the Board of Directors since inception and as President since June 2010. Dr. Lederman has been the Chairman of Krele since its inception in August 2010. Since 1996, Dr. Lederman has been an Associate Professor at Columbia University. As an Assistant Professor at Columbia, Dr. Lederman discovered and characterized the CD40-ligand and invented therapeutic candidates to treat autoimmune diseases and transplant rejection. Dr. Lederman has been a Manager of L&L Technologies LLC since 1996. In addition, Dr. Lederman has been the Managing Member of Seth Lederman Co, LLC since January 2007 and the Managing Member of Lederman & Co, LLC since 2002, both of which are biopharmaceutical consulting and investing companies. Dr. Lederman has also been the Managing Member of Targent Pharmaceuticals since 2000, and Managing Member of Plumbline LLC since 2002. Targent Pharmaceuticals, LLC was a founder of Targent Pharmaceuticals Inc. on which Board of Directors Dr. Lederman served from inception in 2001 until the sale of its assets to Spectrum Pharmaceuticals Inc. in 2006. Between January 2007 and November 2008, Dr. Lederman was a Managing Partner of Konanda Pharma Partners, LLC, a Director of Konanda Pharma Fund I, LP, and a Managing Partner of Konanda General Partner, LLC, which were related private growth equity fund entities. As well, between January 2007 and November 2008, Dr. Lederman was Chairman of Validus Pharmaceuticals, Inc. and Fontus Pharmaceuticals, Inc., which were portfolio companies of the Konanda private growth equity fund. Since December 2011, Dr. Lederman has served as CEO and Chairman of Leder Laboratories Inc. and Starling Pharmaceuticals Inc, which are biopharmaceutical development companies. Between 2006 and 2011, Dr. Lederman was a director of Research Corporation, a New York-based non-profit Dr. Lederman received his BA degree in Chemistry from Princeton University in 1979 and his MD from Columbia University in 1983. Dr. Lederman has been a New York State licensed physician since 1985. Dr. Lederman’s significant experience with our patent portfolio and his experience as an entrepreneur, seed capital investor, fund manager, and director of start-up biopharmaceutical companies were instrumental in his selection as a member of the board of directors.
Benjamin Selzer became our Chief Operating Officer in October 2011 and our interim Chief Financial Officer, Secretary and Treasurer in February 2012. Mr. Selzer has served as the Chief Operating Officer of Tonix Sub since April 2011. Between February 2011 and April 2011, Mr. Selzer served as Tonix Sub’s Chief Business Officer. Between May 2009 and January 2011, Mr. Selzer was a private consultant. Previously, Mr. Selzer was the Executive Director, International Operations and Alliance Management at Aton Pharma, Inc. from April 2008 to May 2009 and Director, Business Development at Reliant Pharmaceuticals, Inc. from July 2004 to March 2008. From 1999 through 2004, Mr. Selzer was a healthcare investment banker at Banc of America Securities LLC, Lehman Brothers Inc., and Warburg Dillon Read LLC in New York. Mr. Selzer received his BA in Economics from The Johns Hopkins University.
Stuart Davidson became a Director in October 2011. Between July 2010 and October 2011, Mr. Davison served as a director of Tonix Sub. Since 1994, Mr. Davidson has been a Managing Partner of Labrador Ventures. Prior to Labrador, Mr. Davidson founded and served as CEO of Combion, Inc., which was acquired by Incyte. He also served as President of Alkermes, Inc., a biotechnology company focused on drug delivery. Mr. Davidson received his Bachelor’s Degree from Harvard College in 1978 and his MBA from Harvard Business School in 1984. Mr. Davidson’s prior experience as a venture capital investor, entrepreneur, and biotechnology industry executive experience leading pharmaceutical companies was instrumental in his selection as a member of our board of directors.
| 48 |
Patrick Grace became a Director in October 2011. Between June 2007 and October 2011, Mr. Grace served as a director of Tonix Sub. Since 1996, he has been a director of Chemed Corporation. Mr. Grace was the co-founder of and has served as the Managing Partner of Apollo Philanthropy Partners, L.L.C. since October 2008. He has also been President of MLP Capital, Inc., New York, New York, an investment holding company, since 1996. Mr. Grace served in various senior management roles with W. R. Grace & Co. from 1977-1995, and was last President and CEO of Grace Logistics Services, Inc. From January 2002 to August 2002, Mr. Grace was also President and Chief Executive Officer of Kingdom Group, LLC (“Kingdom”), New York, New York (a provider of turnkey compressed natural gas fueling systems), which filed for bankruptcy January 2002, and he was Executive Vice President of Kingdom from August 1999 to December 2000. Mr. Grace was a liberal arts major at the University of Notre Dame and earned a MBA in finance from Columbia University. Mr. Grace’s extensive executive experience, along with his membership on the board of directors of a public company was instrumental in his selection as a member of our board of directors.
Donald W. Landry, MD, PhD became a Director in October 2011. Between June 2007 and October 2011, Dr. Landry served as a director of Tonix Sub. Dr. Landry has been a member of the faculty of Columbia University since 1986, and has served as the Samuel Bard Professor of Medicine, Chair of the Department of Medicine and Physician-in-Chief at New York Presbyterian Hospital/Columbia University since 2008. Dr. Landry was a co-founder and has been a member of L&L Technologies, LLC since 1996. Dr. Landry received his BS degree in Chemistry from Lafayette College in 1975, his PhD in Organic Chemistry from Harvard University in 1979 and his M.D. from Columbia University in 1983. Dr. Landry has been a New York State licensed physician since 1985. In 2008, Dr. Landry was awarded the Presidential Citizens Medal, the second-highest award that the President can confer upon a civilian. Dr. Landry’s significant medical and scientific background was instrumental in his selection as a member of the board of directors.
Ernest Mario, PhD became a Director in October 2011. Between September 2010 and October 2011, Dr. Mario served as a director of Tonix Sub. Dr. Mario is a former Deputy Chairman and Chief Executive of Glaxo Holdings plc and a former Chairman and Chief Executive Officer of ALZA Corporation. Since August 2007, Dr. Mario has served as a Director of Celgene Corporation, a Director of Boston Scientific since October 2001 and currently is the Lead Director of Pharmaceutical Product Development, Inc. From 2003 to 2007, he was Chairman and Chief Executive of Reliant Pharmaceuticals, Inc. Since August 2007, Dr. Mario has served as the Chief Executive Officer and Chairman of Capnia, Inc., a privately held specialty pharmaceutical company in Palo Alto, CA. He is Chairman of the American Foundation for Pharmaceutical Education and serves as an advisor to the pharmacy schools at the University of Rhode Island and The Ernest Mario School of Pharmacy at Rutgers University. In 2007, Dr. Mario was awarded the Remington Medal by the American Pharmacists’ Association, pharmacy’s highest honor. Dr. Mario received a PhD and an MS in physical sciences from the University of Rhode Island and a BS in pharmacy from Rutgers University. Dr. Mario brings to his service as a director his significant executive leadership experience, including his experience leading several pharmaceutical companies, as well as his membership on public company boards and foundations. He also has extensive experience in financial and operations management, risk oversight, and quality and business strategy.
Charles Mather IV became a Director in October 2011. Between April and October 2011, Mr. Mather served as a director of Tonix Sub. Mr. Mather has been the Head of Private and Alternative Capital and Co-Head of ECM at Janney Montgomery Scott since December 2009. Between October 2008 and December 2009, Mr. Mather served as an independent consultant to various securities firms. Between May 2007 and September 2008, Mr. Mather was the head of the Structured Equity Group at Jefferies Group Inc. Prior to that, Mr. Mather held various senior investment banking positions at Cowen and Company, including as Co-Head of the Private Equity Group. Mr. Mather’s extensive experience as an investment banker was instrumental in his selection as a member of our board of directors.
John Rhodes became a Director in October 2011. Between October 2010 and October 2011, Mr. Rhodes served as a director of Tonix Sub. Mr. Rhodes has been a director of Dewey Electronics Company, a manufacturer of electronic and electromechanical systems for the military and commercial markets, since 2005. Between April 2007 and June 2010, Mr. Rhodes was a Senior Advisor to Good Energies, Inc., a renewable energy company. Mr. Rhodes is a former Vice President of Booz Allen Hamilton, Inc. Mr. Rhodes is a graduate of Princeton University and the Yale School of Management. Mr. Rhodes’ extensive business and consulting experience, along with his membership on the board of directors of a public company was instrumental in his selection as a member of our board of directors.
Family Relationships
None.
| 49 |
Board Independence
We are not required to have any independent members of the Board of Directors. The board of directors has determined that (i) Seth Lederman, has a relationship which, in the opinion of the board of directors, would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and is not an “independent director” as defined in the Marketplace Rules of The NASDAQ Stock Market and (ii) Stuart Davidson, Patrick Grace, Donald Landry, Ernest Mario, Charles Mather and John Rhodes are each an independent director as defined in the Marketplace Rules of The NASDAQ Stock Market.
Meetings and Committees of the Board of Directors
During the fiscal year ended December 31, 2011, our board of directors held two meetings and approved certain actions by unanimous written consent. We currently have one board committee, the audit committee. The board as a whole carries out the functions of the compensation and nominating committees. We expect our directors to attend all board and committee meetings and to spend the time needed and meet as frequently as necessary to properly discharge their responsibilities.
Audit Committee
Our Audit Committee consists of Patrick Grace and Charles Mather, with Mr. Grace elected as Chairman of the Committee. Our Board of Directors has determined that each of Messrs. Grace and Mather are “independent” as that term is defined under applicable SEC rules and under the current listing standards of the NASDAQ Stock Market. Mr. Grace is our audit committee financial expert.
Our Audit Committee’s responsibilities include: (i) reviewing the independence, qualifications, services, fees, and performance of the independent auditors, (ii) appointing, replacing and discharging the independent auditor, (iii) pre-approving the professional services provided by the independent auditor, (iv) reviewing the scope of the annual audit and reports and recommendations submitted by the independent auditor, and (v) reviewing our financial reporting and accounting policies, including any significant changes, with management and the independent auditor. The Audit Committee has reviewed and discussed with management the Company’s audited financial statements for the year ended December 31, 2011. Based on the reviews and discussions referred to above, the Audit Committee has recommended to the Board of Directors that the financial statements referred to above be included in this Form 10-K.
Involvement in Certain Legal Proceedings
Our Directors and Executive Officers have not been involved in any of the following events during the past ten years:
| 1. | any bankruptcy petition filed by or against such person or any business of which such person was a general partner or executive officer either at the time of the bankruptcy or within two years prior to that time; | |
| 2. | any conviction in a criminal proceeding or being subject to a pending criminal proceeding (excluding traffic violations and other minor offenses); | |
| 3. |
being subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction, permanently or temporarily enjoining him from or otherwise limiting his involvement in any type of business, securities or banking activities or to be associated with any person practicing in banking or securities activities; | |
|
|
4. | being found by a court of competent jurisdiction in a civil action, the Securities and Exchange Commission or the Commodity Futures Trading Commission to have violated a federal or state securities or commodities law, and the judgment has not been reversed, suspended, or vacated; |
| 5. | being subject of, or a party to, any federal or state judicial or administrative order, judgment decree, or finding, not subsequently reversed, suspended or vacated, relating to an alleged violation of any federal or state securities or commodities law or regulation, any law or regulation respecting financial institutions or insurance companies, or any law or regulation prohibiting mail or wire fraud or fraud in connection with any business entity; or | |
| 6. | being subject of or party to any sanction or order, not subsequently reversed, suspended, or vacated, of any self-regulatory organization, any registered entity or any equivalent exchange, association, entity or organization that has disciplinary authority over its members or persons associated with a member. |
| 50 |
Section 16(a) Beneficial Owner Reporting Compliance
Since we are governed under Section 15(d) of the Exchange Act, we are not required to file reports of executive officers and directors and persons who own more than 10% of a registered class of our equity securities pursuant to Section 16(a) of the Exchange Act.
Code of Ethics
We have adopted a Code of Ethics and Business Conduct that applies to all of our directors, officers and employees. A copy of the Code of Ethics is incorporated by reference as an exhibit.
| 51 |
ITEM 11 - EXECUTIVE COMPENSATION
Summary Compensation Table
The following table provides certain summary information concerning compensation awarded to, earned by or paid to our Chief Executive Officer, the two highest paid executive officers and up to two other highest paid individuals whose total annual salary and bonus exceeded $100,000 for fiscal years 2011 and 2010.
| Name & Principal
Position | Year | Salary ($) | Bonus ($) | Stock Awards ($) | Option Awards ($) | Non-Equity Incentive Plan Compensation ($) | Change in Pension Value and Non- Qualified Deferred Compensation Earnings ($) | All Other Compensation ($) | Total ($) | |||||||||||||||||||||||||||
| Seth Lederman (1) | 2011 | - | - | - | - | - | - | 300,750 | (3) | 300,750 | ||||||||||||||||||||||||||
| Chief Executive Officer | 2010 | - | - | 69,738 | (2) | - | - | - | 205,833 | (3) | 275,571 | |||||||||||||||||||||||||
| Rhonda Rosen (4) | 2011 | 140,463 | - | - | - | - | - | - | 140,463 | |||||||||||||||||||||||||||
| Chief Financial Officer | 2010 | 93,750 | - | 8,865 | (5) | - | - | - | - | 102,615 | ||||||||||||||||||||||||||
| David J. Moss (6) | 2011 | - | - | - | - | - | - | - | - | |||||||||||||||||||||||||||
| Chief Executive Officer | 2010 | - | - | - | - | - | - | - | - | |||||||||||||||||||||||||||
| Mark Lawson (7) | 2010 | - | - | - | - | - | - | - | - | |||||||||||||||||||||||||||
| Chief Executive Officer | ||||||||||||||||||||||||||||||||||||
| Robert Gebert (8) | 2010 | - | - | - | - | - | - | - | - | |||||||||||||||||||||||||||
| Chief Executive Officer | ||||||||||||||||||||||||||||||||||||
| Susan Oliver (9) | 2011 | 113,249 | - | - | - | - | - | - | 113,249 | |||||||||||||||||||||||||||
| Secretary | ||||||||||||||||||||||||||||||||||||
| (1) | Dr. Lederman become our President and Chief Executive Officer on October 7, 2011. His compensation reflects payments made to him either through Tonix or Tonix Sub. |
| (2) | Represents (i) 52,362 shares of common stock granted to Lederman & Co., LLC, and (ii) 256,575 shares of common stock granted to L&L Technologies, LLC, which stock was vested at a value of $0.23/share as of December 31, 2010. |
| (3) | Represents $96,000 and $56,000 of consulting fees paid to L&L Technologies, $198,750 and $145,833 of consulting fees paid to Lederman & Co. and $6,000 and $4,000 of director fees paid for the years ended December 31, 2011 and 2010, respectively. |
| (4) | Ms. Rosen become our Chief Financial Officer on October 7, 2011. Her compensation reflects payments made to her either through Tonix or Tonix Sub. Ms. Rosen was terminated effective February 16, 2012. |
| (5) | Represents 39,272 shares of common stock granted and vested at a value of $0.23/share as of December 31, 2010. |
| (6) | Mr. Moss become our Chief Executive Officer on November 22, 2010 and resigned effective October 7, 2011. |
| (7) | Mr. Lawson become our Chief Executive Officer on January 14, 2010 and resigned on November 22, 2010. |
| (8) | Mr. Gebert resigned as our Chief Executive Officer on January 14, 2010. |
| (9) | Ms Oliver was terminated effective October 20, 2011. |
Option/SAR Grants in Fiscal Year Ended December 31, 2011
None.
Outstanding Equity Awards at Fiscal Year-End Table
None.
| 52 |
Equity Compensation Plan Information
| Plan category | Number of securities to be issued upon exercise of outstanding options (a) | Weighted- average exercise price of outstanding options (b) | Securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) (c) | |||||||||
| Equity compensation plans approved by security holders | - | $ | - | 4,000,000 | ||||||||
| Equity compensation plans not approved by security holders | - | - | - | |||||||||
| Total | - | $ | - | 4,000,000 | ||||||||
Employment Contracts and Termination of Employment and Change-In-Control Arrangements
On April 1, 2011, Tonix Sub entered into an employment agreement with Mr. Selzer, pursuant to which Mr. Selzer was engaged to serve as the Chief Operating Officer of Tonix Sub. Pursuant to this agreement, as amended, Mr. Selzer earns a salary of $175,000 per annum. Mr. Selzer’s salary shall increase to $250,000 on October 7, 2012.
Director Compensation
The following table sets forth summary information concerning the total compensation paid to our non-employee directors in 2011 for services to our company.
| Name | Fees Earned or Paid in Cash ($) | Stock Awards ($) | Total ($) | |||||
| Stuart Davidson | 6,000 | - | 6,000 | |||||
| Patrick Grace | 4,000 | - | 4,000 | |||||
| Donald Landry | 6,000 | - | 6,000 | |||||
| Ernest Mario | 6,000 | - | 6,000 | |||||
| Charles Mather IV | 6,000 | - | 6,000 | |||||
| John Rhodes | 6,000 | - | 6,000 | |||||
| Total: | 34,000 | - | 34,000 | |||||
| 53 |
ITEM 12- SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The following table sets forth certain information regarding beneficial ownership of our common stock as of March 20, 2012:
| • | by each person who is known by us to beneficially own more than 5% of our common stock; | |
| • | by each of our officers and directors; and | |
| • | by all of our officers and directors as a group. |
Unless otherwise indicated in the footnotes to the following table, each person named in the table has sole voting and investment power and that person’s address is c/o Tonix Pharmaceuticals Holding Corp., 509 Madison Avenue, Suite 306, New York New York 10022.
| NAME OF OWNER | TITLE OF CLASS |
NUMBER OF SHARES OWNED (1) |
PERCENTAGE OF COMMON STOCK (2) |
|||||
| Seth Lederman | Common Stock | 12,375,744 | (3) | 35.55 | % | |||
| Benjamin Selzer | Common Stock | 532,350 | 1.55 | % | ||||
| Stuart Davidson | Common Stock | 1,387,288 | (4) | 4.04 | % | |||
| Patrick Grace | Common Stock | 130,906 | * | |||||
| Donald Landry | Common Stock | 2,377,177 | (5) | 6.92 | % | |||
| Ernest Mario | Common Stock | 1,212,745 | 3.53 | % | ||||
| Charles Mather IV | Common Stock | 120,369 | * | |||||
| John Rhodes | Common Stock | 950.936 | 2.77 | % | ||||
| Officers and Directors as a Group (8 persons) | Common Stock | 15,248,001 | (6) | 43.34 | % | |||
| Lederman & Co., LLC (7) | Common Stock | 6,051,765 | (8) | 17.56 | % | |||
| Eli Lederman (9) | Common Stock | 2,401,810 | (10) | 6.99 | % | |||
| L&L Technologies, LLC (11) | Common Stock | 1,934,657 | (12) | 5.64 | % | |||
| National Holdings Corporation (13) | Common Stock | 1,865,406 | 5.44 | % | ||||
| David J. Moss (14) | Common Stock | 2,229,518 | (15) | 6.44 | % | |||
* Denotes less than 1%
(1) Beneficial Ownership is determined in accordance with the rules of the SEC and generally includes voting or investment power with respect to securities. Shares of common stock subject to options or warrants currently exercisable or convertible, or exercisable or convertible within 60 days of March 20, 2011 are deemed outstanding for computing the percentage of the person holding such option or warrant but are not deemed outstanding for computing the percentage of any other person.
(2) Percentage based upon 34,278,432 shares of common stock issued and outstanding as of March 20, 2012.
(3) Includes 5,873,565 shares of common stock and 178,200 shares of common stock underlying warrants owned by Lederman & Co., LLC, 1,904,857 shares of common stock and 29,800 shares of common stock underlying warrants owned by L&L Technologies, Inc., 1,179,424 shares of common stock and 326,700 shares of common stock underlying warrants owned by Targent Pharmaceuticals, LLC and 73,961 shares owned by the Seth M. Lederman 1999 Trust. Seth Lederman, as the Managing Member of Lederman & Co., LLC and Targent Pharmaceuticals, LLC, the Manager of L&L Technologies, Inc. and the Trustee of the Seth M. Lederman 1999 Trust, has investment and voting control over the shares held by these entities.
(4) Includes 1,090,882 shares of common stock and 99,000 shares of common stock underlying warrants owned by Lysander, LLC and 130,906 shares owned by Oystercatcher Trust. Stuart Davidson, as the Member of Lysander, LLC and Trustee of Oystercatcher Trust, has investment and voting control over the shares held by these entities.
| 54 |
(5) Includes 1,904,857 shares of common stock and 29,800 shares of common stock underlying warrants owned by L&L Technologies, Inc. Donald Landry, as a Member of L&L Technologies, Inc., has investment and voting control over the shares held by this entity.
(6) Includes 5,873,565 shares of common stock and 178,200 shares of common stock underlying warrants owned by Lederman & Co., LLC, 1,904,857 shares of common stock and 29,800 shares of common stock underlying warrants owned by L&L Technologies, Inc., 1,179,424 shares of common stock and 326,700 shares of common stock underlying warrants owned by Targent Pharmaceuticals, LLC, 73,961 shares owned by the Seth M. Lederman 1999 Trust, 1,090,882 shares of common stock and 99,000 shares of common stock underlying warrants owned by Lysander, LLC and 130,906 shares owned by Oystercatcher Trust.
(7) Seth Lederman, our President and Chief Executive Officer, has investment and voting control over the shares held by this entity. The mailing address for this entity is 245 E. 93rd St. 14E, New York, New York 10128.
(8) Includes 178,200 shares of common stock underlying warrants.
(9) The mailing address for this beneficial owner is Malt House Cottage, Hurley, Berkshire, SL6 5LT, United Kingdom.
(10) Includes 99,000 shares of common stock underlying warrants.
(11) Seth Lederman, our President and Chief Executive Officer and Donald Landry, a Director, have investment and voting control over the shares held by this entity. The mailing address for this entity is 245 E. 93rd St. 14E, New York, New York 10128.
(12) Includes 29,800 shares of common stock underlying warrants.
(13) Mark Goldwasser, C.E.O. has investment and voting control over the shares held by this entity. The mailing address for this entity is 120 Broadway, 27th Floor, New York, NY 10271.
(14) The mailing address for this beneficial owner is 23046 Avenida de la Carlota, Suite 600, Laguna Hills, CA 92653.
(15) Includes 342,700 shares of common stock underlying warrants.
| 55 |
ITEM 13 - CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
Other than as disclosed below, during the last two fiscal years, there have been no transactions, or proposed transactions, which have materially affected or will materially affect us in which any director, executive officer or beneficial holder of more than 5% of the outstanding common, or any of their respective relatives, spouses, associates or affiliates, has had or will have any direct or material indirect interest. We have no policy regarding entering into transactions with affiliated parties.
On June 4, 2010, Tonix Sub entered into a consulting agreement with Lederman & Co., LLC, of which our Chairman, CEO and President Seth Lederman is the Managing Member. Pursuant to this agreement, Lederman & Co. shall provide clinical development, strategic, management and operational consulting services. In exchange for its services, Tonix Sub shall pay Lederman & Co. compensation of $250,000 per annum and issued to Lederman & Co. 261,784 shares of its common stock, 20% of which vested on the date of the agreement and the remainder vesting in equal amounts on each of the first, second, third and fourth anniversaries of the date of the agreement. On August 1, 2011, the cash compensation was reduced to $127,000 per annum. On February 1, 2012, the cash compensation was increased to $250,000 per annum. Immediately prior to the Share Exchange, the unvested shares of common stock vested.
On June 4, 2010, Tonix Sub entered into a technology transfer and assignment agreement with Lederman & Co., LLC. Pursuant to this agreement, Lederman & Co. transferred intellectual property rights related to isometheptene mucate to Tonix Sub. In exchange for the assignment of the intellectual property rights, Tonix Sub issued to Lederman & Co. 1,308,921 shares of its common stock.
On June 4, 2010 Tonix Sub entered into a consulting agreement with L&L Technologies, LLC, of which our Chairman, CEO and President Seth Lederman is the Manager. Pursuant to this agreement, L&L Technologies shall provide scientific and medical consulting services. In exchange for its services, Tonix Sub shall pay L&L Technologies compensation of $96,000 per annum, or such greater amount as the Board may designate from time to time, and issued to L&L Technologies 1,026,194 shares of its common stock, 25% of which vested on the date of the agreement and the remainder vesting in equal amounts on each of the first, second and third anniversaries of the date of the agreement. Immediately prior to the Share Exchange, the unvested shares of common stock vested.
ITEM 14 – PRINCIPAL ACCOUNTING FEES AND SERVICES
Audit Fees. The aggregate fees billed by our independent auditors, for professional services rendered for the audit of our annual financial statements for the year ended December 31, 2011, including review of our interim financial statements were $140,000. Audit fees in respect of 2010 financial statements were $55,000.
Audit Related Fees. We incurred fees to our independent auditors of $80,333 for audit related fees during the fiscal years ended December 31, 2011, which relates to filings with the SEC related to our recent reverse merger, and $-0- for 2010.
Tax and Other Fees. We incurred fees to our independent auditors of $-0- for tax and fees during the fiscal years ended December 31, 2011 and 2010.
The Audit Committee pre-approves all auditing services and all permitted non-auditing services (including the fees and terms thereof) to be performed by our independent registered public accounting firm.
| 56 |
PART IV
ITEM 15 – EXHIBITS, FINANCIAL STATEMENT SCHEDULES
Exhibits:
| 2.01 | Share Exchange Agreement, dated as of October 7, 2011 by and among Tamandare Explorations Inc., David J. Moss, Tonix Pharmaceuticals, Inc. and the shareholders of Tonix Pharmaceuticals, Inc. filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 3.01 | Articles of Incorporation, filed as an exhibit to the Registration Statement on Form S-1, filed with the Securities and Exchange Commission (the “Commission”) on April 9, 2008 and incorporated herein by reference. |
| 3.02 | Articles of Merger between Tamandare Explorations Inc. and Tonix Pharmaceuticals Holding Corp., effective October 11, 2011, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 17, 2011 and incorporated herein by reference. |
| 3.03 | Amended and Restated Bylaws, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on February 23, 2012 and incorporated herein by reference. |
| 10.01 | Feasibility and Option Agreement, dated as of June 20, 2007, by and between Krele Pharmaceuticals, Inc. (now, Tonix Pharmaceuticals, Inc.) and Lipocine, Inc., filed as an exhibit to the amended Current Report on Form 8-K/A, filed with the Commission on February 3, 2012 and incorporated herein by reference. † |
| 10.02 | Consulting Agreement, dated as of June 4, 2010, by and between Krele Pharmaceuticals, Inc. (now, Tonix Pharmaceuticals, Inc.) and Lederman & Co., LLC, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 10.03 | Technology Transfer and Assignment Agreement, dated as of June 4, 2010, by and between Krele Pharmaceuticals, Inc. (now, Tonix Pharmaceuticals, Inc.) and Lederman & Co., LLC, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 10.04 | Lease Agreement, dated as of September 28, 2010, by and between 509 Madison Avenue Associates, L.P. and Tonix Pharmaceuticals, Inc., filed as an exhibit to the amended Current Report on Form 8-K/A, filed with the Commission on February 3, 2012 and incorporated herein by reference. |
| 10.05 | Amendment to Feasibility and Option Agreement, dated as of October 4, 2010, by and between Tonix Pharmaceuticals, Inc. and Lipocine, Inc., filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. † |
| 10.06 | Engagement Agreement, dated as of October 6, 2010, by and between Tonix Pharmaceuticals, Inc. and Frost and Sullivan, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 10.07 | Amendment to Consulting Agreement, dated as of December 9, 2010, by and between Tonix Pharmaceuticals, Inc. and Lederman & Co., LLC, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 10.08 | Employment Agreement, dated as of April 1, 2011, by and between Tonix Pharmaceuticals, Inc. and Rhonda Rosen, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 10.09 | Employment Agreement, dated as of April 1, 2011, by and between Tonix Pharmaceuticals, Inc. and Benjamin A. Selzer, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 57 |
| 10.11 | Employment Agreement, dated as of April 1, 2011, by and between Tonix Pharmaceuticals, Inc. and Susan Oliver (now, Susan Kerridge), filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 10.11 | API Supply and Development Agreement, dated as of April 7, 2011, by and between Tonix Pharmaceuticals, Inc. and JFC Technologies, Inc., filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 10.12 | Consulting Agreement, dated as of June 2, 2011, by and between Tonix Pharmaceuticals, Inc. and Pharmanet Canada, Inc., filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 10.13 | Amendment to Employment Agreement, dated as of July 27, 2011, by and between Tonix Pharmaceuticals, Inc. and Rhonda Rosen, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 10.14 | Amendment to Employment Agreement, dated as of July 27, 2011, by and between Tonix Pharmaceuticals, Inc. and Benjamin A. Selzer, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 10.15 | Amendment to Employment Agreement, dated as of July 27, 2011, by and between Tonix Pharmaceuticals, Inc. and Susan Oliver (now, Susan Kerridge), filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 10.16 | Financial Public Relations Agreement, dated as of August 1, 2011, by and between Tonix Pharmaceuticals, Inc. and Porter, LeVay & Rose, Inc., filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 10.17 | Form of 8% Secured Convertible Debenture, issued October 7, 2011, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 10.18 | Form of Subscription Agreement, dated October 7, 2011, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 10.19 | Form of Pledge and Security Agreement, dated as of October 7, 2011, by and among Tamandare Explorations Inc., Tonix Pharmaceuticals, Inc., Krele LLC and the investors, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 10.20 | Form of Subsidiary Guaranty, dated as of October 7, 2011, by and among Tonix Pharmaceuticals, Inc., Krele LLC and Sandor Capital Master Fund L.P., on behalf of the investors, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 10.21 | Form of Subscription Agreement, dated January 20, 2012, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on January 23, 2012 and incorporated herein by reference. |
| 10.22 | Form of Class A Warrant, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on January 23, 2012 and incorporated herein by reference. |
| 10.23 | Form of Class B Warrant, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on January 23, 2012 and incorporated herein by reference. |
| 10.24 | Form of Registration Rights Agreement, dated January 20, 2012, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on January 23, 2012 and incorporated herein by reference. |
| 10.25 | Amendment to Consulting Agreement, dated as of March 30, 2012 but effective as of July 27, 2011, by and between Tonix Pharmaceuticals, Inc. and Lederman & Co., LLC |
| 14.01 | Code of Ethics and Business Conduct for Officers, Directors and Employees, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on February 23, 2012 and incorporated herein by reference. |
| 58 |
| 21.01 | List of Subsidiaries, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 31.01 | Certification of Chief Executive Officer pursuant to Exchange Act Rules 13a-14(a) and 15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
| 31.02 | Certification of Chief Financial Officer pursuant to Exchange Act Rules 13a-14(a) and 15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
| 32.01 | Certifications of Chief Executive Officer and Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
| 99.01 | Frost & Sullivan Fibromyalgia Market Study, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 99.02 | Lipocine Cyclobenzaprine Study Results, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 14, 2011 and incorporated herein by reference. |
| 101 INS | XBRL Instance Document* |
| 101 SCH | XBRL Schema Document* |
| 101 CAL | XBRL Calculation Linkbase Document* |
| 101 LAB | XBRL Labels Linkbase Document* |
| 101 PRE | XBRL Presentation Linkbase Document* |
| 101 DEF | XBRL Definition Linkbase Document* |
† Confidential treatment is requested for certain confidential portions of this exhibit pursuant to Rule 24b-2 under the Exchange Act. In accordance with Rule 24b-2, these confidential portions have been omitted from this exhibit and filed separately with the Commission.
* The XBRL related information in Exhibit 101 shall not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, or otherwise subject to liability of that section and shall not be incorporated by reference into any filing or other document pursuant to the Securities Act of 1933, as amended, except as shall be expressly set forth by specific reference in such filing or document.
| 59 |
SIGNATURES
In accordance with the requirements of the Exchange Act, the registrant caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| TONIX PHARMACEUTICALS HOLDING CORP. | |||
| Date: March 30, 2012 | By: | /s/ SETH LEDERMAN | |
| Seth Lederman | |||
| Chief Executive Officer (Principal
Executive Officer) |
|||
| Date: March 30, 2012 | By: | /s/ BENJAMIN SELZER | |
| Benjamin Selzer | |||
| Chief Financial Officer (Principal
Accounting Officer) |
|||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Name | Position | Date | ||
| /s/ SETH LEDERMAN | Director | March 30, 2012 | ||
| Seth Lederman | ||||
| Director | March 30, 2012 | |||
| Stuart Davidson | ||||
| /s/ PATRICK GRACE | Director | March 30, 2012 | ||
| Patrick Grace | ||||
| /s/ DONALD W. LANDRY | Director | March 30, 2012 | ||
| Donald W. Landry | ||||
| /s/ ERNEST MARIO | Director | March 30, 2012 | ||
| Ernest Mario | ||||
| /s/ CHARLES MATHER IV | Director | March 30, 2012 | ||
| Charles Mather IV | ||||
| /s/ JOHN RHODES | Director | March 30, 2012 | ||
| John Rhodes |
| 60 |