UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM
(Mark One)
For
the Fiscal Year Ended
Commission
File Number
(Exact name of registrant as specified in its charter)
| (State or other jurisdiction of incorporation or organization) | (IRS Employer Identification No.) | |
| (Address of principal executive office) | (Zip Code) | |
(Registrant’s telephone number, including area code) |
||
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol | Name of each exchange on which registered | ||
Securities registered pursuant to Section 12(g) of the Act: None
Indicate
by check mark if the registrant is a well-known seasoned issuer, as defined by Rule 405 of the Securities Act. Yes ☐
Indicate
by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ☐
Indicate
by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange
Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports),
and (2) has been subject to such filing requirements for the past 90 days.
Indicate
by check mark whether the registrant has submitted electronically, if any, every Interactive Data File required to be submitted
pursuant to Rule 405 of Regulation S-T (§ 229.405 of this chapter) during the preceding 12 months (or for such shorter period
that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and an “emerging growth company” in Rule 12b-2 of the Exchange Act
| Large accelerated filer ☐ | Accelerated filer ☐ |
| Smaller
reporting company | |
| Emerging
growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate
by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness
of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered
public accounting firm that prepared or issued its audit report.
If
securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the
registrant included in the filing reflect the correction of an error to previously issued financial statements.
n
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate
by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐
The
aggregate market value of the voting common equity held by non-affiliates as of June 30, 2025, based on the closing sales price
of the common stock as quoted on The NASDAQ Stock Market was $
As of March 11, 2026, there were shares of registrant’s common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
TABLE OF CONTENTS
2
PART I
ITEM 1 – BUSINESS
This Annual Report on Form 10-K (including the section regarding Management’s Discussion and Analysis of Financial Condition and Results of Operations) contains forward-looking statements regarding our business, financial condition, results of operations and prospects. All statements contained in this Annual Report other than statements of historical fact, including, but not limited to, statements regarding the launch and commercialization of TONMYA and our migraine products, our future results of operations and financial position, business strategy, market size, potential growth opportunities, clinical and nonclinical development activities, efficacy and safety profile of our marketed products and product candidates, potential therapeutic benefits and economic value of TONMYA™, our migraine products and our product candidates, our ability to market and sell our products while maintaining full compliance with applicable federal and state laws, rules and regulations, the timing and results of nonclinical studies and clinical trials, the potential impact of global business or macroeconomic conditions, including as a result of inflation, changing interest rates, cybersecurity incidents, significant political, trade or regulatory developments and global regional conflicts on our operations, and the receipt and timing of potential regulatory designations, approvals and commercialization of product candidates. Words such as “expects,” “anticipates,” “intends,” “plans,” “believes,” “seeks,” “estimates” and similar expressions or variations of such words are intended to identify forward-looking statements, but are not deemed to represent an all-inclusive means of identifying forward-looking statements as denoted in this Annual Report on Form 10-K. Additionally, statements concerning future matters are forward-looking statements.
Although forward-looking statements in this Annual Report on Form 10-K reflect the good faith judgment of our management, such statements can only be based on facts and factors currently known by us. Consequently, forward-looking statements are inherently subject to risks and uncertainties and actual results and outcomes may differ materially from the results and outcomes discussed in or anticipated by the forward-looking statements. Factors that could cause or contribute to such differences in results and outcomes include, without limitation, those specifically addressed under the heading “Risks Factors” below, as well as those discussed elsewhere in this Annual Report on Form 10-K. Readers are urged not to place undue reliance on these forward-looking statements, which speak only as of the date of this Annual Report on Form 10-K. We file reports with the Securities and Exchange Commission (“SEC”). You can read and copy any materials we file or will file with the SEC, which, among other places, can be found on the SEC’s website at http://www.sec.gov, as well as on our corporate website at www.tonixpharma.com).
We undertake no obligation to revise or update any forward-looking statements in order to reflect any event or circumstance that may arise after the date of this Annual Report on Form 10-K. Readers are urged to carefully review and consider the various disclosures made throughout the entirety of this Annual Report, which attempt to advise interested parties of the risks and factors that may affect our business, financial condition, results of operations and prospects.
3
Business Overview
We (“Tonix” or the “Company”) are a fully-integrated biopharmaceutical company commercializing and developing innovative therapies for central nervous system (“CNS”) disorders, immunology, infectious diseases, and rare diseases. Our portfolio consists of commercial, development and discovery-stage programs. In August 2025, we received approval from the U.S. Food and Drug Administration (“FDA”) for TONMYA for the treatment of fibromyalgia in adults. TONMYA is our first internally developed product to receive FDA approval. We hold worldwide commercialization rights to TONMYA, a centrally acting, non-opioid analgesic designed for bedtime administration and long-term use.
We launched TONMYA™ (cyclobenzaprine HCl sublingual tablets) for the treatment of fibromyalgia in adults on November 17, 2025. TONMYA is the first new medicine for fibromyalgia in more than 15 years. In addition to TONMYA, we market two FDA-approved prescription products for the treatment of acute migraine: Zembrace® SymTouch® (sumatriptan injection) and Tosymra® (sumatriptan nasal spray). Our commercial platform includes sales, marketing, market access, distribution, and patient support capabilities. We have generated a diversified pipeline of development candidates through internal discovery, in-licensing, acquisitions, and collaborations with, commercial, academic and non-profit institutions. With commercial operations established and a broad development portfolio, our strategy is to grow TONMYA into a leading therapy for fibromyalgia, advance pipeline programs, and pursue strategic business development opportunities.
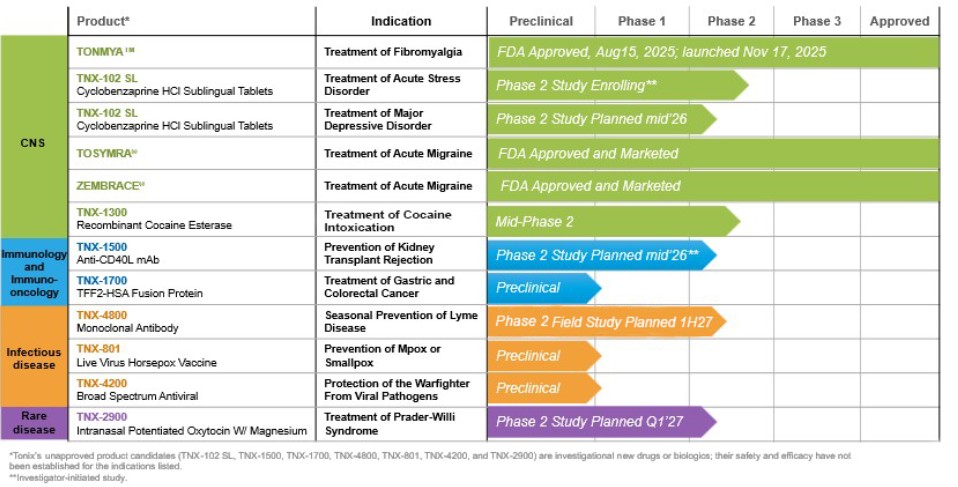
Our development pipeline includes multiple programs in clinical and preclinical development.
The proprietary cyclobenzaprine HCl sublingual tablet formulation contained in TONMYA is referred to as “TNX-102 SL” outside of the fibromyalgia indication. We are exploring the utility of TNX-102 SL (cyclobenzaprine HCl sublingual tablets) in Phase 2 clinical trials for major depressive disorder (“MDD”) and acute stress disorder (“ASD”) and acute stress reaction (“ASR”). TNX-102 SL is being developed to treat ASR and ASD under an Investigator-Initiated investigational new drug application (“IND”) at the University of North Carolina (“UNC”) in the ongoing OASIS study for which UNC received funding from the U.S. Department of Defense (“DoD”). A Phase 2 study of TNX-102 SL for MDD is expected to commence mid-2026 under a Tonix IND that has been cleared by the FDA.
Our clinical stage programs also include:
| ● | Prevention of Lyme Disease: TNX-4800 (anti-OspA from Borrelia burgdorferi), a monoclonal antibody for seasonal prevention of Lyme disease, for which initiation of a Phase 2 field study is planned for the first half of 2027 and of a Phase 2 human challenge study for 2028, pending FDA clearances. |
| ● | Prevention of Kidney Transplant Rejection/Treatment of Autoimmune Diseases: TNX-1500, a humanized monoclonal antibody targeting CD40-ligand (CD40L or CD154), which is Phase 2-ready and being developed for the prevention of kidney transplant rejection and for which an investigator-initiated Phase 2 study in kidney transplant is expected to initiate in mid-2026. TNX-1500 is also in development for the treatment of autoimmune diseases. |
| ● | Treatment of Cocaine Intoxication: TNX-1300, a double-mutant cocaine esterase, which is in Phase 2 for the treatment of cocaine intoxication. TNX-1300 has been granted Breakthrough Therapy designation by the FDA. |
| ● | Treatment of Prader-Willi Syndrome: TNX-2900, intranasal oxytocin potentiated with magnesium, in development for Prader-Willi syndrome and initiation of a Phase 2 study is planned for the first quarter of 2027. TNX-2900 has been granted Orphan Drug designation and Rare Pediatric Disease designation. |
| ● | Treatment of Acute Stress Disorder and Acute Stress Reaction: TNX 102-SL – Collaboration with University of North Carolina, Investigator-initiated IND, currently enrolling Phase 2 study. |
| ● | Treatment of Major Depressive Disorder: TNX 102-SL – Planning to initiate a potentially pivotal Phase 2 study mid-2026. |
Our pre-clinical programs include:
| ● | Vaccine for Protection against mpox and smallpox: TNX-801 (horsepox, live virus vaccine) for mpox and smallpox. TNX-801 is expected to enter a Phase 1 study in 2027 pending FDA clearance. TNX-801 is in the pre-IND stages of development. |
| ● | Broad Spectrum Anti-viral to protect against viral diseases: TNX-4200, a small molecule broad-spectrum antiviral agent targeting CD45 for the prevention or treatment of high lethality infections to improve the medical readiness of military personnel in biological threat environments. The TNX-4200 program is supported by an up to $34 million contract over five years from the DoD’s Defense Threat Reduction Agency (“DTRA”). TNX-4200 is in the pre-IND stages of development. |
| ● | Treatment of gastric and colorectal cancer: TNX-1700, a fusion protein of TFF2 and albumin, is in preclinical development for the treatment of gastric and colorectal cancer in combination with PD-1 blockade, in-licensed from Columbia University. TNX-1700 is in the pre-IND stages of development. |
| ● | Non-opioid analgesic for neuropathic pain: TNX-4900, a highly selective small-molecule Sigma-1 receptor (“S1R”) antagonist, for neuropathic pain, in the pre-IND stages of development. |
With commercial operations established and a broad development portfolio advancing, our strategy is to grow TONMYA into a leading therapy for fibromyalgia, advance pipeline programs, and pursue strategic business development opportunities.
Our commercial portfolio of FDA-approved medicines includes:
TONMYA (cyclobenzaprine HCl sublingual tablets) – Fibromyalgia
TONMYA, a proprietary sublingual formulation of cyclobenzaprine designed for bedtime dosing and long-term use, is approved in the United States for the treatment of fibromyalgia in adults and was launched in November 2025. TONMYA was approved under the 505(b)(2) pathway based on three Phase 3 studies, including two studies that demonstrated statistically significant improvement in pain compared with placebo. Clinical studies showed rapid onset of benefit, sustained efficacy, and a safety profile consistent with known cyclobenzaprine effects, with the most common adverse events being transient local oral reactions.
4
TONMYA is designed to target the non-restorative sleep that we believe is central to fibromyalgia pathophysiology. TONMYA’s sublingual formulation is designed for transmucosal delivery to bypass first-pass hepatic metabolism, which reduces formation of norcyclobenzaprine, the persistent active metabolite that we believe interferes with the duration of any treatment effects from oral, swallowed cyclobenzaprine. The sublingual, transmucosal formulation results in a distinct pharmacokinetic profile compared to oral cyclobenzaprine, with greater relative bioavailability of the parent drug and reduced active metabolite exposure during sleep.
We commercialize TONMYA through our sales organization, which includes an internal sales force, and a contracted salesforce, non-personal promotion, digital engagement, market access programs, patient support services, and distribution through national wholesalers and specialty distributors. We are focused on obtaining payer coverage, building physician awareness, and driving adoption primarily in practices that have a history of diagnosing and treating fibromyalgia, which includes rheumatology, primary care, pain management and neurology practices.
We have contracted for two commercial supply sources of TONMYA, one of which is Almac Pharma Services, a member of the privately owned Almac Group.
We are pursuing lifecycle management strategies for TONMYA, including potential label expansion, real-world evidence generation, and geographic expansion.
Zembrace® SymTouch® (sumatriptan injection) and Tosymra® (sumatriptan nasal spray) - Migraine Franchise
Zembrace SymTouch (sumatriptan injection) 3 mg is a low-dose autoinjector designed for ease of use and rapid onset of action. Zembrace is the only branded sumatriptan autoinjector actively promoted in the United States and has patent protection into 2036. Tosymra (sumatriptan nasal spray) 10 mg is a rapid-acting intranasal formulation using Intravail® permeation enhancer technology with patent protection into 2031. Effective January 1, 2026, Tosymra has preferred exclusive placement on a payer formulary representing approximately 16 million covered lives.
Zembrace SymTouch and Tosymra are both indicated for the treatment of acute migraine with or without aura in adults. Zembrace SymTouch is the only branded sumatriptan autoinjector professionally promoted in the United States and is designed for ease of use and favorable tolerability with a low 3 mg dose. Tosymra is a novel intranasal sumatriptan product formulated with a permeation enhancer that provides rapid and efficient absorption of sumatriptan. Tosymra was approved on the basis of bioequivalence to subcutaneous (s.c.) sumatriptan. Tonix Medicines is the only manufacturer with both a branded injectable and nasal spray indicated for the acute treatment of migraine with or without aura in adults.
Development Pipeline
Central Nervous System
In September 2025, we announced the successful completion of a Type B Pre-IND meeting with the FDA regarding the development of TNX-102 SL for the treatment of MDD. We received positive feedback from the FDA and plan to pursue a supplemental new drug application (“sNDA”) to expand the therapeutic indication of TNX-102 SL to include MDD, based on exploratory findings suggesting that improving sleep quality may positively impact depressive symptoms.
In November 2025, we announced the FDA cleared the IND application to support clinical development of TNX-102 SL 5.6 mg for the treatment of MDD in adults. The unique pharmacological profile of TNX-102 SL is designed to target the disturbed sleep which is often associated with depression. Prior studies of TNX-102 SL in fibromyalgia and post-traumatic stress disorder (“PTSD”) showed promising signals for improvement of depressive symptoms on the Beck Depression Inventory-II and the Montgomery-Åsberg Depression Rating Scale (“MADRS”), respectively.
The IND clearance enables Tonix to proceed with a potentially pivotal Phase 2 HORIZON study, a 6-week, randomized, double-blind, placebo-controlled study of TNX-102 SL as a first-line monotherapy in adults with MDD. About 360 patients will be enrolled at approximately 30 U.S. sites. Eligible participants are 18 years or older and currently experiencing a moderate to severe major depressive episode. The study will compare TNX-102 SL 5.6 mg, taken sublingually at bedtime, to placebo, with the primary endpoint being the MADRS total score change from baseline at Week 6. Secondary endpoints include global impression scores, anxiety ratings, and measures of sleep disturbance. We plan to initiate enrollment of the study in mid-year 2026.
5
TNX-102 SL also is being developed as a treatment for ASR and ASD under an investigator-initiated IND with the University of North Carolina Institute for Trauma Recovery.
In addition, TNX-102 SL has active INDs for the treatment of PTSD, agitation in Alzheimer’s disease (“AAD”), alcohol use disorder (“AUD”) and the management of multi-site pain associated with Long COVID (also known as Post-Acute SARS-CoV-2 or PASC). TNX-102 SL for AAD has been granted Fast Track designation by the FDA. We are currently not actively studying TNX-102 SL in PTSD, AAD, Long COVID or AUD, and are continuously evaluating further indications for which TNX-102 SL could potentially provide benefit.
We are also developing TNX-1300 (double-mutant cocaine esterase), which is in Phase 2 for the treatment of cocaine intoxication. TNX-1300 has been granted Breakthrough Therapy designation by the FDA. TNX-1300 was licensed from Columbia University in 2019 after a Phase 2 study showed that it rapidly and efficiently disintegrates cocaine in the blood of volunteers who received intravenous (i.v.) cocaine. We received a Federal Grant from the U.S. National Institute on Drug Abuse (“NIDA”), a part of the U.S. National Institutes of Health (“NIH”), to advance the development of TNX-1300 as a treatment for cocaine intoxication, and the funding period is now completed. Because of the challenges of recruiting eligible patients into a Phase 2 study, we terminated that study and intend to meet with the FDA in 2026 to inform the clinical design of our next Phase 2 study.
We are developing TNX-1900 (intranasal potentiated oxytocin) for several CNS disorders through investigator-initiated studies. TNX-1900 is in development through investigator-initiated studies at Massachusetts General Hospital (“MGH”) for the treatment of binge eating disorder (“BED”), adolescent obesity, bone health in pediatric autism, and arginine-vasopressin deficiency.
Finally, in December 2025, we in-licensed TNX-4900, formerly known as PW507, from Rutgers University. TNX-4900 is a highly selective S1R antagonist with demonstrated analgesic activity in multiple models of neuropathic pain. TNX-4900 is in the pre-IND stages of development.
Immunology and Infectious Disease
Our lead candidate in the infectious disease pipeline is TNX-4800. Tonix in-licensed worldwide rights to TNX-4800 (formerly known as mAb 2217LS), in September 2025. TNX-4800 is a long-acting fully human monoclonal antibody that targets the outer surface protein A (OspA) of Borrelia burgdorferi, the causative agent of Lyme disease in humans. TNX-4800 is being developed for annual seasonal use and was invented and developed by researchers at UMass Chan Medical School, which has licensed the technology to Tonix. There are currently no FDA-approved vaccines or prophylactics to protect against Lyme disease.
TNX-4800 has an engineered extended half-life and targets the outer-surface protein A (OspA) on Lyme-causing Borrelia bacteria. By binding OspA when TNX-4800 containing blood is ingested by the tick, TNX-4800 kills and blocks the maturation of Borrelia burgdorferi in the mid-gut of infected deer ticks. Published work in non-human primates showed that TNX-4800 was 95% effective in preventing infection after 6 days of exposure to ticks infected with Borrelia burgdorferi. TNX-4800 was derived from mAb 2217 by amino acid substitutions in its crystallizable fragment (Fc) domain which served to prolong the serum half-life. A single administration in the Spring is designed to provide onset of immunity within two days and maintain protective antibody titers for the entire tick season, providing pre-exposure prophylaxis against Lyme disease without relying on the recipient’s immune system to generate antibodies. By delivering a well-characterized antibody directly, TNX-4800 has been shown to block transmission of the major Borrelia genospecies from ticks to animals. TNX-4800 also sidesteps the elaborate immunization schedules required for OspA vaccines in development and an FDA-approved vaccine that was withdrawn from the market. Tonix intends to advance TNX-4800 through additional clinical trials with the goal of submitting a Biologics Licensing Application (BLA) to the FDA.
We intend to meet with the FDA in 2026 to explore Phase 2/3 development options for TNX-4800, including a Phase 2 field study and a Phase 2 human challenge study. The proposed field study would test TNX-4800 for prevention of Lyme in volunteers at risk for Lyme in regions of the U.S. where Lyme is endemic. The proposed challenge study – also called a controlled human infection model (“CHIM”) study – would test the ability of TNX-4800 to protect against Borrelia infection by exposing treated or control volunteers to Borrelia-infected ticks that mimic natural infection. Pending clearances from FDA, we plan to initiate the Phase 2 field study in 2027 and the Phase 2 human challenge study in 2028. Production of investigational product under GMP is underway to enable initiation of the field early in 2027, pending FDA clearance. In animals, TNX-4800 provides protection in approximately two days against the bacteria that causes Lyme disease after a single administration. The passive immunity conferred by TNX-4800 is very different from the active immunity conferred by vaccines in development to protect against Lyme disease in that TNX-4800 kills and blocks the metamorphic-like transformation of Borrelia in the tick’s midgut, preventing transmission of the bacteria, whereas vaccines illicit an immune response in the body of a vaccinated person. Prophylaxis with TNX-4800 may also mitigate some of the limitations of vaccine products designed to actively immunize against Lyme, including suboptimal immune responses due to age, immunocompetence, and other reasons.
6
Within immunology, we are developing TNX-1500, an Fc-modified humanized mAb, directed against CD40-ligand (CD40L, also known as CD154). TNX-1500 was engineered to modulate binding to Fc receptors with the goal of maintaining the activity of first-generation mAbs, yet with reduced risk of thrombotic complications. TNX-1500 is being developed to prevent organ transplant rejection as well as to treat autoimmune conditions. Topline results from a Phase 1 single ascending dose escalation study at 3 mg/kg, 10 mg/kg and 30 mg/kg of TNX-1500 in healthy volunteers were reported in the first quarter of 2025. Pharmacokinetic results showed mean half-life (t1/2) for the 10 mg/kg and 30 mg/kg dose groups of 34-38 days, consistent with monthly dosing. In healthy volunteers, TNX-1500 was generally well-tolerated with a favorable safety profile. Anti-CD40L has multiple potential indications in addition to solid organ and bone marrow transplantation including autoimmune diseases. In November 2025, we announced an investigator-initiated study with MGH, a founding member of Mass General Brigham (“MGB”), to conduct a Phase 2 clinical trial evaluating TNX-1500 in kidney transplant recipients. The investigator-initiated study will be led by Ayman Al Jurdi, M.D., at MGH and is designed to assess the safety, tolerability and activity of Fc-modified anti-CD40L mAb TNX-1500 in preventing kidney transplant rejection while significantly minimizing the dose of conventional immunosuppressive drugs, which are associated with infection, cancer, cardiovascular side effects and various metabolic derangements. The study is expected to be initiated mid-year 2026 pending FDA clearance of the IND.
Our immunology pipeline also includes TNX-1700, a recombinant Trefoil Factor Family 2 fused to human serum albumin (“hTFF2-HSA”) that was licensed from Columbia University in 2019. TNX-1700 is an immunotherapy being developed to treat gastric and colorectal cancers, in combination with PD-1 blockers, and is at the preclinical stage of development. Results of preclinical testing demonstrated that a mouse version of TNX-1700 was able to evoke an increase in anti-tumor immunity in combination with anti-PD-1 in several mouse models of gastric cancer by reducing immunosuppressive neutrophils and activating anti-tumoral CD8+ T cell responses. TNX-1700 as both monotherapy and in combination with anti-PD-1 was also able to dramatically reduce metastasis and increase survival in these models. TNX-1700 also exhibits efficacy in various mouse models of colorectal cancer in combination with anti-PD-1. An INTERACT (INitial Targeted Engagement for Regulatory Advice on CBER/CDER ProducTs) meeting was held with the FDA in 2025 and received constructive early guidance on program development.
Our infectious disease portfolio includes vaccines based on our live virus vaccine or recombinant pox vaccine (“RPV”) platform. Live virus vaccines are believed to protect against poor clinical outcomes of infectious diseases by eliciting T-cell responses in addition to antibody responses. TNX-801, a live minimally replicative vaccine based on synthesized horsepox, is in the pre-IND stage of development to protect against smallpox and mpox. Preclinical data/studies demonstrate that TNX-801, regardless of route of administration (i.e. intradermal, subcutaneous or intramuscular), provided 100% protection against vaccinia virus and monkeypox virus challenge in terms of both mortality and clinical disease (lesions) in a highly sensitive rabbitpox model.
TNX-801 also serves as the live virus vaccine platform for other infectious diseases, for which subsequent products will be designed by expressing other viral antigens in the horsepox vector. Our GMP-capable advanced manufacturing facility in Dartmouth, Massachusetts was purpose-built to manufacture TNX-801. The GMP suites are currently decommissioned and may be reactivated on the earlier of 2027 or in the case of a national or international emergency.
We are developing a potential broad-spectrum antiviral CD45-targeted therapeutic (TNX-4200). The DoD announced in December 2022 a plan to move beyond a “one bug, one drug” approach and is seeking broad-spectrum drugs as it may be hard to predict which or how many viruses may be deployed on the battlefield.
In July 2024, we were awarded a contract with a potential for up to $34 million over five years by DTRA. The objective of the contract is to develop small molecule broad-spectrum antiviral agents for the prevention or treatment of infections to improve the medical readiness of military personnel in biological threat environments. The program focuses on optimization and development of TNX-4200 to develop an orally available CD45 antagonist with broad-spectrum efficacy against a range of viral families through preclinical evaluation. The program is expected to establish physicochemical properties, pharmacokinetics, and safety attributes to support an IND submission and to fund a first-in-human Phase 1 clinical study. Tonix plans to leverage previous research on phosphatase inhibitors, specifically compounds that target CD45, to optimize lead compounds for therapeutic intervention of biothreat agents and provide the government with a complete and cost-effective solution for a broad-spectrum medical countermeasure. We believe that partial inhibition of CD45 will provide optimal antiviral protection while requiring lower plasma drug concentrations, a lower dose, and a better safety profile.
We will utilize our state-of-the-art research laboratory capabilities, including a Biosafety Level 3 (BSL-3) lab and Animal Biosafety Level 3 (ABSL-3) facility, in Frederick, Maryland (“RDC”), as well as experienced in-house personnel, to develop vaccines and antiviral therapies for mpox, smallpox and other infectious diseases. We intend to collaborate with academic partners to test the efficacy of CD45 inhibitor compounds against multiple viral select agents using BSL-4 facilities.
7
Rare Disease Pipeline
Our rare disease portfolio consists of TNX-2900 (intranasal potentiated oxytocin) for Prader-Willi syndrome (“PWS”), a rare genetic disorder and the leading cause of life-threatening childhood obesity, affecting about 1 in 10,000 to 1 in 30,000 births. Infants often present with poor muscle tone and feeding difficulties, while children and adolescents develop hyperphagia, behavioral challenges, and severe obesity and metabolic disease. Current interventions are difficult to sustain and often inadequate. The formulation technology for TNX-2900 was acquired from Trigemina, Inc. and licensed from Stanford University in 2020. The potentiated formulation includes magnesium, which has been shown in animal studies to potentiate binding of oxytocin to the oxytocin receptor. The therapeutic technology was licensed from Inserm, the French National Institute of Health and Medical Research. TNX-2900 was granted Orphan-Drug Designation by the FDA in the second half of 2023, and the IND was cleared by the FDA in the fourth quarter of 2023, and received Rare Pediatric Disease Designation in March 2024, which would make us eligible for a transferable Priority Review Voucher upon approval.
We plan to progress our TNX-2900 program for the treatment of PWS into a Phase 2, randomized, double-blind, placebo-controlled, parallel-design study to evaluate the safety, tolerability, and efficacy of TNX-2900 in male and female participants with PWS, ages 8 to 17.5 years. Eligible participants will be randomized to receive 12-weeks of treatment with TNX-2900 at one of three dose levels, or placebo, in a 1:1:1:1 ratio. The primary efficacy endpoint will be the change from baseline in the validated Hyperphagia Questionnaire for Clinical Trials (HQ-CT), a widely used measure of hyperphagia severity in PWS. Secondary objectives will include assessments of behavior, caregiver burden, and quality of life measures, as well as safety and tolerability outcomes. We intend to initiate enrollment in this study in the first quarter of 2027.
Facilities
Relating to our development programs, we own and operate the RDC in Frederick, Maryland consisting of one building totaling approximately 48,000 square feet. The RDC conducts research on CNS, immunology, and infectious disease candidates. The RDC facility is mostly biosafety level 2 (BSL-2), with some components designated BSL-3. We also own an Advanced Development Center (“ADC”) located in the New Bedford business park in Dartmouth, Massachusetts. This approximately 45,000 square foot BSL-2 facility is intended to accelerate development, clinical and commercial scale manufacturing of live-virus vaccines and biologics to support clinical trials. This facility was decommissioned in 2024, and may be reactivated on the earlier of 2027 or in the case of a national or international emergency.
We are led by a management team with significant industry experience in commercialization and drug development. We complement our management team with a network of scientific, clinical, and regulatory advisors that includes recognized experts in their respective fields.
Our Strategy
Our strategy is to use our integrated development and marketing capabilities to advance innovative programs across multiple therapeutic areas through the drug development process, with the ultimate objectives of FDA approval and commercialization. The principal components of our strategy are to:
●
|
Drive the successful commercialization of TONMYA for the treatment of fibromyalgia in the United States. TONMYA was approved by the FDA in August 2025 for the treatment of fibromyalgia in adults. We launched TONMYA in the United States in November 2025 and an important objective is to establish TONMYA as the standard of care for fibromyalgia. We have approximately 90 U.S. sales representatives who are focused on promoting TONMYA to physicians and other healthcare prescribers who treat fibromyalgia, including rheumatologists, primary care physicians, pain specialists, neurologists and psychiatrists.
| |
| ● | Maximize the commercial potential of our product candidates. We plan to commercialize our product candidates, either on our own or through collaboration with partners. Alternatively, we could enter into partnership agreements with drug companies that already have significant marketing capabilities in the same, or similar, therapeutic areas. |
|
● | Pursue additional indications and commercial opportunities for our product candidates. We plan to maximize the value of certain of our products and product candidates by pursuing other indications and commercial opportunities for such candidates. For example, we are exploring the development and commercialization of TNX-102 SL for MDD, ASD/ASR and other important indications. For TNX-1900, we are exploring the development for the treatment of binge eating disorder, adolescent obesity, bone health in pediatric autism, and arginine-vasopressin deficiency. Finally, our live virus platform using our RPV technology may be developed as vaccines for future pandemics, infectious diseases generally, in addition to smallpox and mpox, and for oncology applications. |
8
| ● | Pursue CNS, rare disease, immunology, and infectious disease indications with high unmet medical need and significant commercial potential. We are pursuing multiple indications that are underserved with limited, effective treatment options. Our broader development strategy is to leverage the patented formulation and proven mechanism of action to explore the clinical potential of TNX-102 SL in multiple other, psychiatric, and addiction conditions, including MDD, ASR and ASD, all of which are underserved by currently approved medications or have no approved treatment. One of our latest stage product candidates is TNX-102 SL for the treatment of MDD, a condition which affects more than 21 million adults in the U.S. While several antidepressant medications are available, many individuals do not achieve adequate relief or discontinue treatment due to side effects like weight gain, sleep disruption, and sexual dysfunction. Cocaine intoxication is one of the leading causes of overdose deaths and for which there is no currently approved therapy, however, those studies are currently on hold. Within CNS, Tonix is also developing TNX-1300 to treat cocaine intoxication and TNX-1900 to treat binge eating disorder, adolescent obesity, bone health in pediatric autism, and arginine-vasopressin deficiency. With TNX-4800, we are pursuing a prevention for Lyme disease, which has the potential to affect millions of people and for which there is no current prophylaxis. With TNX-1500, we are pursuing a treatment to prevent organ transplant rejection as well as autoimmune conditions. TNX-1500 is a third generation humanized mAb targeting CD40L that has the potential to deliver efficacy without compromising safety, based on modulated binding to Fc receptors. At this time, no mAb against CD40L has been licensed anywhere in the world. Within infectious diseases, we are also focusing on the development of TNX-801 to prevent smallpox and mpox. While there are FDA-approved vaccines to prevent smallpox and mpox, we believe TNX-801 has potential to provide durable protection. | |
| ● | Pursue a broad intellectual property strategy to protect our product candidates. We are pursuing a broad patent strategy for our product candidates, and we endeavor to generate new patent applications as supported by our innovations and conceptions as well as to advance their prosecution. In the case of TONMYA, we own patents and patent applications protecting its composition-of-matter, certain methods of its use, its formulation, and its pharmacokinetic properties. We plan to opportunistically apply for new patents to protect our product candidates. |
Disease and Market Overview
Our product candidates address disorders that are not well served by currently available therapies or have no approved treatment which represent large potential commercial market opportunities. Background information on the disorders and related commercial markets that may be addressed by our product candidates in or nearing the clinical stage of development is set forth below.
Central Nervous System
Fibromyalgia (FM)
Fibromyalgia is a common chronic pain disorder that is understood to result from amplified sensory and pain signaling within the central nervous system, called central sensitization. Brain imaging studies have localized the functional disorder to the brain’s insular and anterior cingulate cortex. Fibromyalgia afflicts more than 10 million adults in the U.S., the majority of whom are women. Symptoms of fibromyalgia include chronic widespread pain, non-restorative sleep, fatigue, and brain fog (or cognitive dysfunction). Other associated symptoms include mood disturbances, including depression, anxiety, headaches, and abdominal pain or cramps. Individuals suffering from fibromyalgia often struggle with their daily activities, have impaired quality of life, and frequently are disabled. Physicians and patients report common dissatisfaction with currently marketed products. Fibromyalgia is now recognized as the prototypic nociplastic syndrome. Nociplastic pain is the third primary type of pain in addition to nociceptive pain and neuropathic pain. Many patients present with pain syndromes that are combinations of the three primary types of pain. Nociplastic syndromes can involve components of both central and peripheral sensitization. Fibromyalgia can occur without any identifiable precipitating event. However, many fibromyalgia cases follow one or more precipitating event(s) including: chronic nociceptive or neuropathic pain states; recovery from an infectious illness; a cancer diagnosis or cancer treatment; a metabolic or endocrine stress; or a traumatic event. In the case of recovery from an infectious illness, fibromyalgia is considered an Infection-Associated Chronic Condition. In addition to fibromyalgia cases associated with other conditions or stressors, the U.S. National Academies of Sciences, Engineering, and Medicine, has concluded that fibromyalgia is a diagnosable condition that occurs after recovery from COVID in the context of Long COVID. Fibromyalgia is also recognized as a Chronic Overlapping Pain Condition, due to shared symptoms with chronic fatigue syndrome/myalgic encephalomyelitis, irritable bowel syndrome, endometriosis, low back pain, post-concussive syndrome (also known as mild traumatic brain injury), chronic Lyme disease, chronic diabetic neuropathy and chronic post-herpetic neuralgia.
9
We believe that diagnosing fibromyalgia in Long COVID patients will increase the potential market for TNX-102 SL as compared to market estimates from before the COVID-19 pandemic. Tonix has previously presented its analysis of real-world evidence from the TriNetX claims database suggesting that over 40% of Long COVID patients present with a constellation of symptoms that overlap with fibromyalgia.
Based on market research which we commissioned, despite the availability of approved medications, the majority of patients fail therapy due to either insufficient efficacy, poor tolerability, or both. Prescription pain and sleep medications, including opioids, are frequently prescribed off-label for symptomatic relief, despite the lack of evidence that such medications provide a meaningful or durable therapeutic benefit, and many of these medications carry significant safety risks and risk of dependence. For example, based on U.S. claims data, approximately 50% of patients diagnosed with FM are prescribed opioids within 18 months of diagnosis, despite the lack of evidence for their effectiveness and the risk of addiction and toxicity, including overdose.
Major Depressive Disorder
MDD is a prevalent and serious psychiatric illness that affects adults of all ages, races, and backgrounds. It is characterized by persistent feelings of sadness or loss of interest, along with symptoms such as sleep and appetite disturbances, fatigue, difficulty concentrating, and thoughts of worthlessness or suicide. These symptoms must last at least two weeks and significantly impair daily functioning. In the United States, more than 21 million adults experience a major depressive episode each year. While several antidepressant medications are available, many individuals do not achieve adequate relief or discontinue treatment due to side effects like weight gain, sleep disruption, and sexual dysfunction. MDD is associated with increased risk of suicide and substantial impairment in quality of life, underscoring the urgent need for new, first-line therapies that are both effective and well-tolerated.
Acute Stress Disorder and Acute Stress Reaction
ASD is a mental health condition that can occur within the first month of experiencing a traumatic event. The symptoms are similar to those of PTSD and can affect both civilian and military populations. ASR is a transient, often severe, emotional and physical response occurring minutes to days after a traumatic event, such as assault, disaster, or accident. Symptoms include anxiety, flashbacks, numbness, and insomnia, typically resolving within a few days or up to one month. According to the National Center for PTSD, in the U.S. about 60% of men and 50% of women experience at least one trauma in their lives. In the U.S. alone, one-third of emergency department visits (40-50 million patients per year) involve evaluation after trauma exposures, and in a 2014 study involving U.S. veterans, 87% reported exposure to at least one potentially traumatic event during their service. No medications are currently available at or near the point of care to treat patients suffering from acute traumatic events and to support long-term health.
Cocaine Intoxication
Cocaine is an illegal recreational drug taken for its pleasurable effects and associated euphoria. Pharmacologically, cocaine blocks the reuptake of the neurotransmitter dopamine from central nervous system synapses, resulting in the accumulation of dopamine within the synapse and an amplification of dopamine signaling that is related to its role in creating positive feeling. With the continued use of cocaine, however, intense cocaine cravings occur resulting in a high potential for abuse and addiction, or dependence, as well as the risk of cocaine intoxication. Cocaine intoxication refers to the deleterious effects on other parts of the body, especially those involving the cardiovascular system. Common symptoms of cocaine intoxication include tachyarrhythmias and elevated blood pressure, either of which can be life-threatening. As a result, individuals with known or suspected cocaine intoxication are sent immediately to the emergency department, preferably by ambulance in case cardiac arrest occurs during transit. There are approximately 505,000 emergency room visits for cocaine abuse each year in the U.S., of which 61,000 require detoxification services. According to the National Institute on Drug Abuse, cocaine-involved deaths rose nearly 54% from 2019 to 2021, resulting in over 24,486 deaths total.
Immunology
Organ Transplant Rejection
Organ transplant rejection occurs when the immune system of the organ recipient attacks the new organ as if it was an infection or tumor. Often transplantation is the last resort for most end-stage organ failure patients, affecting either kidneys, liver, heart, lungs, and/or pancreas. Genetic disparity between organ donor and recipient is often at the root of the rejection. Mismatched or not closely matched organs trigger an immune reaction that leads to rejection. Overcoming this difficulty is paramount to a patient’s survival as organ donations are in limited supply.
10
Gastric and Colorectal Cancers
Gastric or stomach cancer is a disease in which malignant cancer cells line the inner lumen of the stomach. Development of this form of cancer is often influenced by age, diet and other stomach diseases. This type of cancer begins to form in the mucosa, the surface of the lumen that is in direct contact with the contents of the stomach, and spreads through the outer layers of the stomach as the tumor grows.
Currently, per the National Cancer Institute, the 5-year relative survival for stomach cancer is 36.4%. According to 2018-2021 data, approximately 0.8 percent of men and women will be diagnosed with stomach cancer during their lifetime. In 2021, there were an estimated 130,263 people living with stomach cancer in the U.S. As of 2024, there were approximately 26,890 new cases with 10,880 deaths.
Colorectal cancer includes cancers in the colon and the rectum, organs that are crucial to absorption of water by the body and the elimination of food-waste. Most colorectal cancers start as a growth or polyp on the inner lining of the colon or rectum. Some types of polyps can change into cancer over time (usually many years), but not all polyps become cancer. Adenomatous polyps are the ones that turn malignant with time. Similar to gastric cancer, malignancy begins in the mucosal layer and spreads outwards.
The 5-year relative survival rate with colorectal cancer is 65.0%, per the National Cancer Institute. Based on 2018-2021 data, approximately 4.0 percent of men and women will be diagnosed with colorectal cancer during their lifetime. In 2021, there were an estimated 1,392,445 people living with colorectal cancer in the United States. As of 2024, there were approximately 152,810 new cases with 53,010 deaths. It is the 3rd leading cause of cancer death in women, and 2nd in men.
Infectious Diseases
Lyme Disease
Lyme disease is caused by the bacterium Borrelia burgdorferi. Lyme disease remains the most common vector-borne infection in the United States and its incidence is climbing each year. It occurs most commonly in the Northeast, mid-Atlantic, and upper-Midwest regions. Lyme disease bacteria are transmitted through the bite of infected Ixodes ticks. Typical symptoms include fever, headache, fatigue, and a characteristic skin rash called erythema migrans. If left untreated, infection can spread to joints, the heart, and the nervous system. Laboratory testing is helpful if used correctly and performed with FDA-cleared tests.
Smallpox and Mpox
Smallpox is an acute contagious disease caused by the variola virus, or VARV, which is a member of the orthopoxvirus family. Smallpox was declared eradicated in 1980 following a global immunization campaign. Smallpox is transmitted from person to person by infective droplets during close contact with infected symptomatic people. Mpox is an acute contagious disease caused by the monkeypox virus or MPXV, which is also a member of the orthopoxvirus family. Mpox symptoms are similar to those of smallpox, although less severe. Mpox is emerging as an important zoonotic infection in humans in Central and West Africa. Until 2022, only a few cases of mpox had been reported outside of Africa in patients who had been infected while in Africa. Starting in May of 2022, mpox clade II cases spread rapidly in the U.S. and other countries. The Clade II mpox affects mostly men who have sex with men in the U.S., where it has become endemic. In August 2024, the World Health Organization (“WHO”) declared mpox Clade Ib to be a public health emergency of international concern (“PHEIC”) due to an outbreak in the Democratic Republic of the Congo that spread globally, including to the United States. Clade Ib affects children as well as adults. Although PHEIC designation has been lifted by WHO, mpox continues to spread in Africa and mutations of the virus are considered by public health experts to be an ongoing threat to be monitored for new epidemic spread.
Smallpox was eradicated by a World Health Organization program that vaccinated individuals with live replicating vaccinia vaccines wherever smallpox appeared. In the 1970s, vaccination of civilians to protect against smallpox was discontinued in the U.S.; however, smallpox remains a material threat to national security and a proportion of military personnel, including members of the Global Response Force continue to be vaccinated. Vaccines for smallpox and mpox are stockpiled by the U.S. government in the strategic national stockpile and for potential widespread immunization in the event of malicious reintroduction of VARV. The U.S. National Academy of Sciences has recently issued a consensus report raising concerns about the state of new mpox vaccines in development.
11
Rare Disease
Prader-Willi Syndrome
PWS is recognized as the most common genetic cause of life-threatening childhood obesity and affects males and females with equal frequency and all races and ethnicities. PWS results from the absence of expression of a group of genes, specifically related to the MAGE (melanoma antigen) gene family on the Prader–Willi critical region (15q11–q13) on the paternally acquired chromosome. The hallmarks of PWS are lack of suckling in newborns and, in children and adolescents, severe hyperphagia – an overriding physiological drive to eat, leading to severe obesity and other complications associated with significant mortality. A systematic review of the morbidity and mortality as a consequence of hyperphagia in PWS found that the average age of death in PWS was 22.1 years. Given the serious or life-threatening manifestations of these conditions, there is a critical need for effective treatments to decrease morbidity and mortality, improve quality of life, and increase life expectancy in people with PWS. Oxytocin has potent effects in correcting behavioral characteristics of the MAGEL2 knock-out mouse model for PWS and autism. Six clinical trials have investigated intranasal oxytocin as a treatment in pediatric patients with PWS. Four clinical studies showed evidence for improvement in PWS-related behaviors/symptoms. Three of these clinical studies reported evidence for improvement in hyperphagia and one showed an improvement in sucking in infants.
Tonix’s Marketed Products
TONMYA – Treatment of Fibromyalgia
In August 2025, we received approval from the FDA for TONMYA (cyclobenzaprine HCl sublingual tablets) for the treatment of fibromyalgia. TONMYA in adults, Tonix’s first internally developed product, was commercially launched by the Company in the United States on November 17, 2025. TONMYA is the first new treatment for fibromyalgia in more than 15 years and is a centrally acting, differentiated non-opioid analgesic designed for bedtime administration and long-term use, addressing core symptoms of fibromyalgia including pain, disturbed sleep and fatigue. The approval and launch of TONMYA marked a major milestone in our evolution as an organization with growing revenues and an expanding customer footprint. We hold worldwide commercialization rights to TONMYA.
Zembrace SymTouch and Tosymra – Acute Migraine in Adults
In June 2023, we acquired two FDA-approved, marketed products from Upsher-Smith: Zembrace SymTouch (sumatriptan injection) 3 mg and Tosymra (sumatriptan nasal spray) 10 mg. Zembrace SymTouch and Tosymra are both indicated for the treatment of acute migraine with or without aura in adults.
Zembrace SymTouch is the only actively promoted brand of sumatriptan autoinjector in the United States. It has a unique low dose and has demonstrated onset of migraine pain relief in as few as 10 minutes (17% of patients vs. 5% for placebo). Zembrace SymTouch also demonstrated migraine pain freedom for 46% of patients (vs 27% for placebo) at 2 hours in a single-attack, double-blind study (N=230). Zembrace SymTouch currently has patent protection to 2036. Tosymra employs Intravail® permeation enhancer technology and is pharmacokinetically equivalent to 4 mg subcutaneous sumatriptan. Tosymra delivers migraine pain relief in as little as 10 minutes with just one spray for some patients (13% vs. 5% for placebo). Tosymra currently has patent protection to 2031.
Together, these products form the foundation of our commercial platform, including sales, marketing, market access, distribution, and patient support capabilities.
Product Candidates in Development
We believe that our product candidates offer innovative therapeutic approaches and may provide significant advantages relative to available therapies. We have worldwide commercialization rights to all of our product candidates listed below. The following table summarizes our later stage product candidates that are in or nearing the clinic:
| Product Candidate | Indication | Stage of Development | ||
| TNX-102 SL | Major Depressive Disorder | Phase 2 expected to commence mid-2026 | ||
| TNX-102 SL | Acute Stress Reaction/Acute Stress Disorder | Phase 2 enrolling* | ||
| TNX-4800 | Seasonal Prevention of Lyme Disease | Phase 2 field study planned first half 2027 and Phase 2 human challenge study planned 2028 (pending FDA clearances) | ||
| TNX-1500 | Kidney Transplant Rejection | Phase 2 study planned for mid 2026* | ||
| TNX-1300 | Cocaine Intoxication | Mid-Phase 2 | ||
| TNX-1900 | Adolescent Obesity, Binge Eating Disorder, Bone Health in Pediatric Autism, and arginine-vasopressin deficiency |
Phase 2 currently enrolling* | ||
| TNX-2900 | Prader-Willi Syndrome | Phase 2 expected to commence first quarter 2027 | ||
| TNX-801 | Smallpox and Mpox vaccine | Preclinical, pre-IND | ||
| TNX-4200 | Treatment or Prevention of Viral Disease | Preclinical, pre-IND | ||
| TNX-4900 | Neuropathic pain | Preclinical, pre-IND | ||
| TNX-1700 | Gastric and colorectal Cancer | Preclinical, pre-IND |
*Investigator Initiated Studies
12
TNX-102 SL
Overview
TNX-102 SL is a proprietary sublingual tablet formulation of cyclobenzaprine (“CBP”) that efficiently delivers CBP across the oral mucosal membrane into the systemic circulation. TNX-102 SL is approved under the brand name TONMYA, for the treatment of fibromyalgia in the U.S. We have active IND’s for TNX-102 SL as a treatment for MDD, ASD/ASR, PTSD, or multi-site pain associated with Long COVID, AAD and AUD however, we are not currently studying TNX-102 SL in PTSD, AAD, Long Covid or AUD. We own all rights to TNX-102 SL in all geographies, and we bear no obligations to third parties for any future development or commercialization. Excipients used in TNX-102 SL are approved for pharmaceutical use. Some of the excipients were specially selected to promote a local oral environment that facilitates transmucosal absorption of CBP.
The current TNX-102 SL sublingual tablets each contain 2.8 mg of CBP. We selected this dose with the goal of providing a balance of efficacy, safety, and tolerability that would be acceptable as a first-line therapy and for long-term use, and in-patient populations characterized by burdensome symptoms and sensitivity to medications.
The active ingredient in TNX-102 SL is CBP, a multi-functional drug that blocks the serotonin-2A, alpha-1 adrenergic, muscarinic M1 and histaminergic H1 receptors.
CBP is a tertiary amine tricyclic, that is the listed active ingredient of two products that are approved in the U.S. for the treatment of muscle spasm: Flexeril® (5 mg and 10 mg oral immediate-release, or IR, tablet) and Amrix® (15 mg and 30 mg oral extended-release capsule or ER capsule). The Flexeril brand of CBP IR tablet has been discontinued since May 2013. There are numerous generic versions of CBP IR tablets on the market. CBP-containing products are approved for short term use (two to three weeks) only as an adjunct to rest and physical therapy for relief of muscle spasm associated with acute, painful musculoskeletal conditions. CBP IR tablets are recommended for three times per day dosing, which results in relatively stable blood levels of CBP after several days of treatment. Extended-release (ER) CBP capsules taken once a day mimic, and flatten, the pharmacokinetic profile of three times per day CBP IR tablets.
Both the IR and ER tablet formulations of CBP result in accumulation of the persistent metabolite norcyclobenzaprine (“norCBP”) to blood levels that exceed the levels of CBP. NorCBP is a secondary amine tricyclic with a relatively stronger inhibitory activity of the norepinephrine transporter (NET) than the parent CBP. We believe that the accumulation of norCBP is undesirable in a medicine to be taken chronically at bedtime because norCBP accumulates over weeks, potentially interfering with the dynamic receptor effects of CBP and also may interrupt sleep quality by inhibiting the NET.
We designed TNX-102 SL to be administered once-daily at bedtime and with the intention for long-term use. We believe the selected dose of TNX-102 SL and its unique pharmacokinetic profile will enable it to achieve a desirable balance of efficacy, safety, and tolerability. Our Phase 1 pharmacokinetic comparative trials showed that, on a dose-adjusted basis, TNX-102 SL results in faster systemic absorption and significantly higher plasma levels of CBP in the first hour following sublingual administration relative to oral IR CBP tablets. It also showed that the sublingual route of administration, which bypasses the “first pass” hepatic metabolism that swallowed medications undergo, results in a higher plasma level of CBP relative to norcyclobenzaprine during sleeping hours when taken at bedtime. We believe the dynamic changes in CBP after TNX-102 SL administration at steady state during chronic use contribute to its activity in treating fibromyalgia. We believe this is the first drug designed to increase the activity of the tertiary amine tricyclic parent and decrease the activity of the secondary amine tricyclic active metabolite. In clinical studies, TNX-102 SL 2.8 mg and TNX-102 SL 5.6 mg were generally well-tolerated, with no drug-related serious and unexpected adverse reactions reported in these studies. The most common adverse event was transient numbness in the mouth after TNX-102 SL administration.
13
Global NDA Requirements
We are planning to develop TNX-102 SL for the treatment of FM in the UK, Europe and Japan. We plan to discuss the development of TNX-102 SL for the treatment of FM with the UK’s Medicines and Healthcare products Regulatory Agency (MHRA), the European Medicines Agency (EMA) and the Japanese Pharmaceuticals and Medical Devices Agency (PMDA). Cyclobenzaprine, the active ingredient of TNX-102 SL, has not been approved in the UK, most countries in Europe, or in Japan. In February 2022, we held an End of Phase 2 Consultation with the Japanese PMDA, to discuss the potential Japan development plan. PMDA has provided guidance on the overall nonclinical package to support a Japan NDA filing for TNX-102 SL for the treatment of FM. We plan to have another Consultation with PMDA to provide program updates and discuss the Japan development plan.
We have also successfully completed a Phase 1 bridging pharmacokinetic study in ethnic Japanese and Chinese volunteers that shows similar characteristics to our historical data in Caucasian volunteers. We believe this will satisfy one of the criteria for approval in Japan and China and will allow us to reference the U.S. efficacy data to support marketing applications in those countries.
A Phase 1 PK study was initiated in March 2022 and the clinical phase was completed in May 2022. Since the similarity in PK profile between people of Japanese and Chinese descent was confirmed, the PK data from the two ethnic groups were pooled as for Asian data (n=20) and compared retrospectively with the Caucasian study data from Study TNX-CY-F110 (n=16). The Asian/Caucasian geometric mean ratios of cyclobenzaprine Cmax, AUC0-T and AUC0-∞ were between 0.9 and 1.11 after both the 5.6 mg dose and the 2.8 mg dose. The 90% CI of Asian/Caucasian geometric mean ratios for Cmax, AUC0-T and AUC0-∞, were all within the formal narrow equivalence limit of 0.8 to 1.25 after both the 5.6 mg dose and 2.8 mg dose, respectively. These results support similarity in cyclobenzaprine PK between Asian (pooled Japanese and Chinese) and Caucasian samples.
TNX-102 SL (cyclobenzaprine HCl sublingual tablets) – Major Depressive Disorder (MDD) Program
We are developing TNX-102 SL as a treatment for MDD. In September 2025, Tonix announced the successful completion of a Type B Pre-IND meeting with the FDA regarding the development of TNX-102 SL for the treatment of MDD. The Company received positive feedback from the FDA and plans to pursue a supplemental new drug application (sNDA) to expand the therapeutic indication of TNX-102 SL to include MDD, based on exploratory findings suggesting that improving sleep quality may positively impact depressive symptoms.
In November 2025, the FDA cleared the IND application to support clinical development of TNX-102 SL 5.6 mg for the treatment of MDD in adults. The unique pharmacological profile of TNX-102 SL is designed to target the disruptive sleep which is often associated with depression. Prior studies of TNX-102 SL in fibromyalgia and post-traumatic stress disorder (PTSD) showed promising signals for improvement of depressive symptoms on the Beck Depression Inventory-II and the MADRS, respectively.
The IND clearance enables Tonix to proceed with a potentially pivotal Phase 2 HORIZON study, a 6-week, randomized, double-blind, placebo-controlled study of TNX-102 SL as a first-line monotherapy in adults with MDD. About 360 patients will be enrolled at approximately 30 U.S. sites. Eligible participants are 18 years or older and currently experiencing a moderate to severe major depressive episode. The study will compare TNX-102 SL 5.6 mg, taken sublingually at bedtime to placebo, with the primary endpoint being the MADRS total score change from baseline at Week 6. Secondary endpoints include global impression scores, anxiety ratings, and measures of sleep disturbance. Tonix plans to initiate enrollment of the study in mid-year 2026.
TNX-102 SL – Acute Stress Disorder Program
TNX-102 SL is being developed as a bedtime treatment for ASR/ASD in collaboration with the University of North Carolina under an investigator-initiated IND.
Phase 2 OASIS Study
This investigator-initiated study is being conducted by the University of North Carolina Institute for Trauma Recovery. The University of North Carolina has been awarded a $3 million grant from the DoD to investigate the potential of Tonix’s TNX-102 SL to reduce the frequency and severity of adverse effects of acute trauma. The proposed Optimizing Acute Stress reaction Interventions with TNX-102 SL (OASIS) trial will examine the safety and efficacy of TNX-102 SL to reduce adverse posttraumatic neuropsychiatric sequelae among patients presenting to the emergency department (“ED”) after a motor vehicle collision. The investigator-initiated trial was commenced in May 2025 and is targeting to enroll approximately 180 individuals who acutely experienced trauma at ED study sites across the U.S. and participants will be randomized in the ED to receive a two-week course of either TNX-102 SL or placebo. The OASIS trial will examine the safety and efficacy of TNX-102 SL to reduce adverse posttraumatic neuropsychiatric sequelae among patients in the ED after a motor vehicle collision. A fourteen-day course of bedtime TNX-102 SL will be tested in the immediate aftermath of motor vehicle collision trauma. The study will test the potential for TNX-102 SL to target trauma-related sleep disturbance and its ability to facilitate recovery from ASR and to prevent PTSD. The results, if positive, may ultimately provide military personnel with a new treatment option that, when administered in the early aftermath of a traumatic event to individuals with ASR symptoms, improves warfighter function.
14
The OASIS trial will build upon a foundation of knowledge and infrastructure developed through the University of North Carolina-led, $40 million AURORA study. The AURORA study is a major national research initiative to improve the understanding, prevention, and recovery of individuals who have experienced a traumatic event. AURORA is supported by funding from the NIH, leading brain health nonprofit One Mind, private foundations, and partnerships with leading tech companies such as Mindstrong Health and Verily Life Sciences, the health care arm of Google’s parent company Alphabet.
We presented clinical data and rationale supporting the potential for TNX-102 SL to be studied for the treatment of ASR and prevention of PTSD. Prior studies showed that treatment with TNX-102 SL showed effects on sleep and PTSD symptoms in PTSD patients at two and four weeks. This supportive data on the effects of TNX-102 SL on reducing PTSD symptoms suggest early intervention immediately after trauma using TNX-102 SL has the potential to reduce ASR and to be prophylactic for development of ASD and PTSD. Data from these trials support testing of TNX-102 SL within 24 hours of index trauma for effects on ASR symptoms and the subsequent incidence of newly developed ASD within one month and PTSD after one month from the index trauma.
TNX-4800 – Lyme Disease Prophylaxis
TNX-4800 (formerly known as mAb 2217LS) is a humanized monoclonal antibody with an engineered extended half-life that targets the outer-surface protein A (OspA) on Lyme-causing Borrelia bacteria. By binding OspA when TNX-4800 containing blood is ingested by the tick, TNX-4800 kills and blocks the maturation of Borrelia burgdorferi in the mid-gut of infected deer ticks. Published work in animals showed that TNX-4800 was 95% effective in preventing infection after 6 days of exposure to ticks infected with Borrelia burgdorferi. TNX-4800 was derived from mAb 2217 by amino acid substitutions in its crystallizable fragment (Fc) domain which served to prolong the serum half-life. A single administration in the Spring is designed to provide onset of immunity within two days and maintain protective antibody titers for the entire tick season, providing pre-exposure prophylaxis against Lyme disease without relying on the recipient’s immune system to generate antibodies. By delivering a well-characterized antibody directly, TNX-4800 has been shown to block transmission of the major Borrelia genospecies from ticks to pre-treated animals. TNX-4800 also sidesteps the multidose schedules required for OspA vaccines in development and the FDA-approved vaccine that was withdrawn from the market. Tonix intends to advance TNX-4800 through additional clinical trials with the goal of submitting a Biologics Licensing Application (BLA) to the FDA.
TNX-4800 was studied in a randomized, double-blind, sequential dose-escalation phase 1 study (NCT04863287) that evaluated safety, tolerability, pharmacokinetics (PK), and immunogenicity of TNX-4800 in healthy adults. Forty-four subjects were randomized and 41 completed the study. Subjects received a single subcutaneous (SC) administration of placebo or TNX-4800 at 0.5, 1.5, 5, or 10 mg/kg. Safety was assessed via clinical and lab evaluations. Drug exposure increased by approximately 25-times for a 20-times increase in dose. Serum TNX-4800 was measurable at the earliest sampling time of 24 hours, indicating rapid systemic absorption. TNX-4800 concentrations remained quantifiable for >200 days in 80% of volunteers at the lowest dose and for up to 350 days in the majority of volunteers at higher doses (i.e., ≥ 1.5 mg/kg). Mean half-life ranged from 62–69 days across groups. Serum concentrations remained quantifiable for up to 12 months in most subjects. Mean exposure for the 10 mg/kg cohort was less than 20% of the highest exposures in a rat toxicology study. Anti-drug antibodies (ADA) were detected in <10% of treated subjects, with no impact on PK. Most adverse events were mild or moderate. TNX-4800 was determined to be generally safe and well tolerated.
15
In infected deer ticks, Borrelia’s OspA lipoprotein binds to tick-gut receptor TROSPA and helps it adhere to the midgut lining. During a tick bite blood meal, Borrelia downregulates OspA, upregulates OspC, and activates motility genes. Borrelia undergoes a metamorphic-like transformation, becoming highly flagellated and mobile, which facilitates migration to the tick salivary glands and invasion of human host tissues. During a tick bite of an animal pre-treated with TNX-4800, the tick ingests host blood containing TNX-4800, which kills and blocks the metamorphic-like transformation of Borrelia in the tick’s midgut preventing transmission of the bacteria. Lyme-causing Borrelia-exposed or -infected individuals, rarely make antibodies against OspA which allows for people to be reinfected despite having immunity to OspC. Consequently, we expect that protection against Borrelia would require annual prophylaxis with TNX-4800.
TNX-1500 – Organ Transplant Rejection/Autoimmune Conditions
TNX-1500 is a humanized mAb directed against CD40-ligand, or CD40L (also known as CD154), engineered to modulate binding to Fc receptors. TNX-1500 is being developed for the prevention of allograft and xenograft rejection, for the prevention of graft-versus-host disease (GvHD) after hematopoietic stem cell transplantation (HCT) and for the treatment of autoimmune diseases. The IND was cleared for the prevention of kidney transplant. TNX-1500 incorporates the antigen binding fragment (Fab) region of hu5c8, which has been extensively characterized including at the atomic level in complex with CD40-ligand.
CD40-ligand is a protein expressed on the surface of activated T lymphocytes that mediates T cell helper function. CD40-ligand is also known as CD154, the T cell-B cell activating molecule (T-BAM), TRAP and gp39. CD154 is a member of the Tumor Necrosis Factor (TNF) Super Family. No mAb against CD154 has been approved for commercial use anywhere in the world. Other TNF Super Family members have been successfully targeted by antagonist mAbs. Approved mAbs against TNFα include: infliximab (Remicade®), adalimumab (Humira®), certolizumab pegol (Cimzia®), and golimumab (Simponi®) for the treatment of certain autoimmune conditions. Also, etanercept (Enbrel®) is a TNFα antagonist receptor fusion protein. An approved mAb against RANKL (CD254) is denosumab (Prolia® or Xgeva®) for the treatment of osteoporosis, treatment-induced bone loss, metastases to bone, and giant cell tumor of bone.
The Fc-modified TNX-1500 has shown activity and has been well tolerated in animals and in a single dose Phase 1 pharmacodynamic (PD) and pharmacokinetic (PK) study that supports monthly dosing. TNX-1500 was engineered to modulate binding to Fc receptors with the goal of maintaining the activity of first-generation monoclonal antibodies (mAbs), yet with reduced risk of thrombotic complications. TNX-1500 is being developed as a prophylaxis against organ transplant rejection as well as to treat autoimmune conditions. A Phase 1 single ascending dose escalation study of TNX-1500 at 3 mg/kg, 10 mg/kg and 30 mg/kg of TNX-1500 in healthy volunteers was initiated in the second quarter of 2023. The objectives of the Phase 1 trial were to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of intravenous TNX-1500, as well as to support dosing in a planned Phase 2 trial in kidney transplant recipients. We reported positive topline data related to the Phase 1 in February of 2025. TNX-1500 showed suppression of the primary and secondary antibody responses to KLH antigen challenges for the 10 and 30 mg/kg doses. Additionally, preliminary pharmacokinetic results showed mean half-life (t1/2) for the 10 mg/kg and 30 mg/kg dose groups of 34-38 days, consistent with monthly dosing. In healthy volunteers, TNX-1500 was generally well-tolerated. Anti-CD40L has multiple potential indications in addition to solid organ and bone marrow transplantation including autoimmune diseases.
In pre-clinical experiments at MGH, TNX-1500 has been studied as monotherapy or in combination with immunosuppressive drugs in heart and kidney organ transplants in animals. The data demonstrates that TNX-1500 showed activity in preventing organ rejection and was well tolerated in animals. Blockade of CD40L with TNX-1500 monotherapy consistently prevented pathologic alloreactive in animal models of cardiac and kidney allograft model without evidence of clinical thrombosis.
In November 2025, we entered into a collaboration with MGH, a founding member of Mass General Brigham (MGB) to conduct an investigator-initiated Phase 2 clinical trial evaluating TNX-1500 in kidney transplant recipients. The study will be led by Ayman Al Jurdi, M.D., at MGH and is designed to assess the safety, tolerability and activity of Fc-modified anti-CD40L mAb TNX-1500 in preventing kidney transplant rejection while significantly minimizing the dose of conventional immunosuppressive drugs, which are associated with infection, cancer, cardiovascular side effects and various metabolic derangements. The CD40 ligand (CD40L) is also known as CD154. Study initiation is contingent on institutional review board (“IRB”) approval and FDA clearance of an investigator-initiated IND. Pending IRB approval and IND clearance, the open-label, single-center study will enroll five adult kidney transplant recipients at MGH. Patients will receive induction therapy with anti-thymocyte globulin, TNX-1500, tacrolimus, and corticosteroids. The corticosteroids will be tapered and discontinued by Day 33 post-transplant. TNX-1500 will be continued for 12 months (to the primary endpoint) with an option to continue treatment beyond 12 months. Tacrolimus at standard dose will be continued for six months, at which point tacrolimus will be decreased to low dose with the expectation of discontinuing tacrolimus after 12 months. The primary endpoint is the incidence of adverse and serious adverse events at 12 months. Secondary endpoints include graft survival, renal function, biopsy-proven acute rejection, and incidence of donor-specific antibodies. The study is expected to be initiated mid-2026.
16
Data published in two peer reviewed articles in the American Journal of Transplantation demonstrate TNX-1500 prevents rejection, prolongs survival and preserves graft function as a single agent or in combination with other drugs in animal renal and heart allografts
TNX-1300 – Cocaine Intoxication
TNX-1300 (T172R/G173Q double-mutant cocaine esterase 200 mg, i.v. solution) is being developed for the treatment of cocaine intoxication. TNX-1300 is a recombinant protein enzyme produced through rDNA technology in a non-disease-producing strain of E. coli bacteria. Cocaine Esterase (“CocE”) was identified in bacteria (Rhodococcus) that use cocaine as the sole source of carbon and nitrogen and that grow in soil surrounding coca plants. The gene encoding CocE was identified and the protein was extensively characterized. CocE catalyzes the breakdown of cocaine into metabolite ecgonine methyl ester and benzoic acid. Wild-type CocE is unstable at body temperature, so targeted mutations were introduced in the CocE gene and resulted in the T172R/G173Q double-mutant CocE, which is active for approximately 6 hours at body temperature.
Currently there is no specific pharmacotherapy indicated for cocaine intoxication, a state characterized by acute agitation, hyperthermia, tachycardia, arrhythmias, and hypertension, with the potential life-threatening sequalae of myocardial infarction, cerebrovascular accident, rhabdomyolysis, respiratory failure, and seizures. Patients are currently managed only by supportive care for the adverse effects of cocaine overdose on the cardiovascular and central nervous systems. By targeting the cause of cocaine intoxication, rather than the symptoms like other medicines in emergency usage, we believe TNX-1300 may offer significant advantages to the current standard of care for cocaine overdose. TNX-1300 was developed by Columbia University, University of Kentucky and University of Michigan, and in-licensed by Tonix from Columbia University in 2019.
In a Phase 2 randomized, double-blind, placebo-controlled clinical study, TNX-1300 at 100 mg or 200 mg i.v. doses was well tolerated and interrupted cocaine effects after cocaine 50 mg i.v. challenge.
In August 2022, we announced that we received a Cooperative Agreement grant from NIDA, part of NIH, to support development of TNX-1300. A positive Phase 2a study of volunteer cocaine users in a controlled laboratory setting has been previously completed. TNX-1300 has been granted Breakthrough Therapy designation by the FDA.
As a biologic and new molecular entity, TNX-1300 is eligible for 12 years of U.S. market exclusivity upon approval by the FDA, in addition to expected patent protection through 2029. Since in-licensing, Tonix has requalified existing inventory, developed a lyophilized drug product to facilitate enhanced stability and handling conditions applicable for an ER treatment, updated the process and analytical methods to current standards and manufactured Phase 2 drug product clinical supply.
We initiated a Phase 2 clinical trial, CATALYST, of TNX-1300 in the third quarter of 2024. The Phase 2 trial was a single-blind, placebo-controlled, proof-of-concept study comparing the safety of a single 200 mg dose of TNX-1300 to standard of care alone in approximately 60 emergency department (ED) patients presenting with cocaine intoxication. Because of the challenges of recruiting eligible patients into a Phase 2 study, we terminated that study and intend to meet with the FDA in 2026 to inform the clinical design of our next Phase 2 study.
TNX-1900 –Adolescent Obesity, Binge Eating Disorder, Bone Health in Pediatric Autism, and Arginine-Vasopressin Deficiency
TNX-1900 (intranasal potentiated oxytocin) is a proprietary formulation of oxytocin primarily in development under investigator-initiated INDs for the treatment of adolescent obesity, binge eating disorder, bone health in pediatric autism, , and arginine-vasopressin deficiency. In 2020, TNX-1900 was acquired from Trigemina, Inc. and licensed from Stanford University. TNX-1900 is a drug-device combination product, based on an intranasal actuator device that delivers oxytocin into the nose.
Oxytocin is a naturally occurring human hormone that acts as a neurotransmitter in the brain. Oxytocin has no recognized addiction potential. It has been observed that low oxytocin levels in the body can lead to an increase in migraine headache frequency, and that increased oxytocin levels can relieve migraine headaches. Certain other chronic pain conditions are also associated with decreased oxytocin levels.
With TNX-1900, the addition of magnesium to the oxytocin formula enhances oxytocin receptor binding as well as its effects on trigeminal neurons and craniofacial analgesic effects in animal models. Intranasal oxytocin has been well tolerated in several clinical trials in both adults and children.
There are four ongoing Phase 2 investigator-initiated studies enrolling at MGH: the POWER study for the treatment of adolescent obesity, the STROBE study for the treatment of binge eating disorder, the BOX study for the treatment of bone health in pediatric autism, and the FOCUS study for the treatment of arginine-vasopressin deficiency.
TNX-2900 – Prader-Willi Syndrome (PWS)
TNX-2900 is based on our patented intranasal potentiated oxytocin formulation, or TNX-1900, but being developed for PWS. We licensed technology using oxytocin-based therapeutics for the treatment of PWS and non-organic failure to thrive disease from Inserm. The licensing agreement has been negotiated and signed by Inserm Transfert, the private subsidiary of Inserm, on behalf of Inserm, Aix-Marseille Université and Centre Hospitalier Universitaire of Toulouse. PWS is recognized as the most common genetic cause of life-threatening childhood obesity and affects males and females with equal frequency and all races and ethnicities. There is currently no approved treatment for either the suckling deficit in infants or the obesity and hyperphagia in older children associated with PWS. Since PWS is an orphan disease that occurs in approximately one in 10,000 to 1 in 30,000 births, TNX-2900 for PWS has been granted Orphan Drug Designation and Rare Pediatric Disease Designation by the FDA. Tonix completed a pre-IND meeting with the FDA in November 2022 to discuss the most efficient and appropriate investigational plan to establish the safety and effectiveness evidence to support the approval of TNX-2900, and Tonix has received IND clearance.
17
The mechanisms involved in suckling activity required for normal feeding and the role of oxytocin system in this process will be investigated. The results of this work are expected to be useful in the clinical care of infants requiring support to achieve efficient suckling behavior. Intranasal oxytocin has previously been shown to improve suckling in newborn animals and suppress feeding behaviors in adult animal models.
Research suggests PWS is associated with a functional deficiency of oxytocin, a neuropeptide that regulates satiety and feeding behaviors through the oxytocin receptor. Oxytocin treatment addresses several key features of PWS expressed in the MAGEL2 (MAGE-like 2) knock-out mouse. Intranasal oxytocin therapy has shown benefits in infants with PWS. Carbetocin has a different spectrum of activity on oxytocin and vasopressin receptors than oxytocin. Oxytocin has dose-related inconsistencies in receptor activity that have been described as “high-dose suppression” or an “inverted “U” dose response. TNX-2900 is formulated with magnesium to further enhance oxytocin receptor binding and signaling, with the goal of providing more consistent and selective receptor activation while minimizing off-target vasopressin effects. In vitro and in vivo in animals Mg++- containing formulations reduce these inconsistencies.
In September 2025, we announced plans to progress its TNX-2900 program for the treatment of PWS into a Phase 2 clinical trial. We plan to conduct a Phase 2 randomized, double-blind, placebo-controlled, parallel-design study to evaluate the safety, tolerability, and efficacy of TNX-2900 in male and female participants with PWS, ages 8 to 17.5 years. Eligible participants will be randomized to receive 12-weeks of treatment with TNX-2900 at one of three dose levels, or placebo, in a 1:1:1:1 ratio. The primary efficacy endpoint will be the change from baseline in the validated Hyperphagia Questionnaire for Clinical Trials (HQ-CT), a widely used measure of hyperphagia severity in PWS. Secondary objectives will include assessments of behavior, caregiver burden, and quality of life measures, as well as safety and tolerability outcomes.
TNX-1700 — Gastric and Colorectal Cancers
TNX-1700 is a recombinant Trefoil Factor Family 2 fused to human serum albumin (“hTFF2-HSA") licensed from The Trustees of Columbia University in the City of New York in development for the treatment of gastric and colorectal cancers. The licensed patents are directed to TFF2 compositions and methods of treatment, U.S. Patent No. 10,124,037 and U.S. Patent No. 11,167,010. The licensed patents provide TNX-1700 with US market exclusivity until April 2033, subject to any patent term extensions. On August 27, 2020, we filed International Patent Application No. PCT/IB2020/000699 entitled “Modified TFF2 Polypeptides.” The PCT application is now nationalized in 12 countries.
In preclinical studies, we have shown efficacy of TNX-1700 in combination with anti-PD-1 in tumor reduction, metastasis and increase in survival in various models of gastric and colorectal cancer. The mechanism of action is to suppress immunosuppressive neutrophils and activate anti-cancer CD8+ T cells, which is distinct from checkpoint inhibitors. There is potential synergy with anti-PD-1 or anti-PD-L1 mAbs.
TNX-801 –Smallpox and Mpox Vaccine
TNX-801 is a novel potential smallpox- and mpox-preventing vaccine based on a synthetic version of live horsepox virus, grown in cell culture. Though it shares structural characteristics with vaccinia-based vaccines, TNX-801 has unique properties that we believe indicate potential safety advantages over existing live replicating vaccinia virus vaccines, which have been associated with adverse side effects such as myopericarditis in some individuals. Emergent BioSolutions’ ACAM2000® is the only replicating vaccinia virus vaccine currently approved by the FDA to protect against smallpox and mpox. We believe replicating virus vaccines have potential efficacy advantages over non-replicating vaccines, relating to the stimulation of cell mediated immunity. Bavarian Nordic’s Jynneos®, the only non-replicating virus vaccine, is currently approved by the FDA to protect against smallpox and mpox. Jynneos® requires two-doses, with an efficacy of approximately 35% after one dose. During the most recent mpox outbreak in the United States, dropout between doses was 24%. We believe TNX-801 has the potential to have improved tolerability relative to replicating vaccinia vaccines and the potential to have improved efficacy relative to non-replicating vaccinia vaccines.
Smallpox was eradicated by a World Health Organization program that vaccinated individuals with live replicating vaccinia vaccines wherever smallpox appeared. In the 1970s, vaccination of civilians to protect against smallpox was discontinued in the U.S.; however, smallpox remains a material threat to national security and a proportion of military personnel, including members of the Global Response Force, continue to be vaccinated. The Bipartisan Commission on Biodefense (2024) noted that “Smallpox and other orthopoxviruses pose significant threats to the United States and the world due to their potential for weaponization, accidental release, and vulnerability of populations who stopped routinely vaccinating against smallpox in the 1970s
We are developing TNX-801 as a potential smallpox- and mpox-preventing vaccine for the U.S. strategic national stockpile and for potential widespread immunization in the event of malicious reintroduction of variola, the virus that causes smallpox.
Mpox has become endemic in the U.S. since it spread in the U.S. and other countries outside of Africa, mostly in populations of gay men. In August 2024, the WHO determined that the upsurge of mpox in a growing number of countries in Africa constitutes a public health emergency of international concern (“PHEIC”), the second such declaration in the past two years in response to transmission of the virus. Mpox cases of the new clade Ib mpox have since also been detected in multiple countries outside of Africa, including the U.S. Animals vaccinated with TNX-801 were protected from mpox in studies reported in the first quarter of 2020. These data were published in the peer-reviewed journal Vaccines in 2023. Although PHEIC designation has been lifted by WHO, mpox continues spread in Africa and mutations of the virus are considered by public health experts to be an ongoing threat to be monitored for new epidemic spread.
In October 2025, at the World Vaccine Congress in Amsterdam, we presented data showing TNX-801, regardless of route of administration (i.e., intradermal, subcutaneous or intramuscular) provided 100% protection against vaccinia virus and monkeypox virus challenge in terms of both mortality and clinical disease (lesions) in a highly sensitive rabbitpox model.
18
In September 2024, at the DoD’s MHSRS conference and in October 2024 at the World Vaccine Congress in Barcelona, Spain, we presented new data on potential mpox vaccine, TNX-801, demonstrating tolerability and no evidence of spreading to blood or tissues, even at high doses, in immunocompromised animals. After a single-dose vaccination, TNX-801 prevented clinical disease and lesions, and also decreased shedding in the mouth and lungs of animals after a lethal challenge with clade Ia monkeypox. These findings are consistent with TNX-801 inducing mucosal immunity and suggest TNX-801 has the ability to block forward transmission. In September 2024, we also announced that the WHO’s preferred TPP aligns with the characteristics of TNX-801. Key elements of the WHO draft TPP include single-dose, durable protection, administration without special equipment, and stability at ambient temperature. Other potential beneficial characteristics include the ability to limit forward transmission, use in case-contact vaccination strategies and suitability for use in immunocompromised individuals.
In October 2023, at the World Vaccine Congress - Europe, we reported that the TNX-801 vaccine was shown to be greater than 10 to 1,000-fold more minimally replicative than older vaccinia-based smallpox vaccines in both human primary cell lines and immunocompromised mice. Similar data was also published in the peer-reviewed journal mSphere which presented data demonstrating that TNX-801 is less virulent than 20th Century vaccinia vaccines in immune-compromised mice.
In August 2023 we received pre-IND meeting written responses from the FDA. Tonix believes the FDA feedback provides a path to agreement on the design of a Phase 1/2 study and the overall clinical development plan. The Phase 1/2 clinical trial will assess the safety, tolerability, and immunogenicity of TNX-801, following the submission and clearance of an IND. We are actively working to develop a vaccine meeting cGMP quality to support a clinical study.
We hold a U.S. Patent for TNX-801 smallpox and mpox vaccine and Recombinant Pox Virus (RPV) platform technology. This patent is expected to provide Tonix with U.S. market exclusivity until 2037, excluding any possible patent term extensions or patent term adjustments. In addition, we expect that TNX-801 will be eligible for 12 years of non-patent-based exclusivity under the Patient Protection and Affordable Care Act, or PPACA.
TNX-801 also serves as the live virus vaccine platform for other infectious diseases for which subsequent products will be designed by expressing other viral antigens in the horsepox vector. Our Good Manufacturing Practice (GMP)-capable advanced manufacturing facility in Dartmouth, MA was purpose-built to manufacture live virus vaccines, including TNX-801. The GMP suites have been decommissioned and may be reactivated the earlier of 2027 or in case of a national or international emergency.
TNX-4200 – Broad-Spectrum Antiviral
We are developing CD45-targeted therapeutics (TNX-4200). In July 2024 Tonix was awarded a contract with a potential for up to $34 million over five years by the U.S. Department of Defense, Defense Threat Agency (DTRA). The objective of the contract is to develop small molecule broad-spectrum antiviral agents for the prevention or treatment of infections to improve the medical readiness of military personnel in biological threat environments.
Our program will focus on optimization and development of its TNX-4200 program, to develop an orally available CD45 antagonist, with broad-spectrum efficacy against a range of viral families through preclinical evaluation. The program is expected to establish physicochemical properties, pharmacokinetics, and safety attributes to support an IND submission and to fund a first-in-human Phase 1 clinical study. We plan to leverage previous research on phosphatase inhibitors, specifically compounds that target CD45, to optimize lead compounds for therapeutic intervention of biothreat agents and provide the government with a complete and cost-effective solution for a broad-spectrum medical countermeasure. Tonix’s hypothesis is that partial inhibition of CD45 will provide optimal antiviral protection while requiring lower plasma drug concentrations, a lower dose, and a better safety profile.
TNX-4900
TNX-4900 is a highly selective S1R antagonist with demonstrated analgesic activity in multiple animal models of neuropathic pain. TNX-4900 was created from a structure-based drug design program led by Dr. Youyi Peng and Dr. William Welsh at Rutgers University that produced a series of potent and selective triazole-based S1R antagonists. The compound binds the human Sigma-1 receptor with nanomolar affinity (Ki = 7.5 nM), demonstrates > 100-fold selectivity over the Sigma-2 receptor, and exhibits high blood-brain barrier penetration and favorable absorption, distribution, metabolism and elimination (ADME) properties, including oral bioavailability of approximately 28%. TNX-4900 is in the pre-IND stages of development. In preclinical models of diabetic and chemotherapy-induced neuropathic pain, TNX-4900 produced significant and durable reductions in pain behaviors after both acute and chronic dosing without evidence of tolerance or motor impairment. Tonix plans to advance TNX-4900 through expanded pharmacokinetic, formulation, and safety studies to support IND-enabling development.
19
Tonix’s Facilities Overview
The Research & Development Center (RDC)
We own the approximately 48,000 square foot RDC facility in Frederick, Maryland. The RDC facility is operational and focuses on our development of vaccines and antiviral drugs against infectious diseases. The RDC is the principal site for the research on TNX-4200, a broad-spectrum antiviral targeting CD45 funded by the DTRA contract. The RDC also conducts research on CNS and immunology drugs. The RDC facility is biosafety level 2 (BSL-2) with BSL-3 components.
The Advanced Development Center (ADC)
The ADC located in the New Bedford business park in Dartmouth, Massachusetts is intended to accelerate development, clinical and commercial scale manufacturing of live-virus vaccines and biologics. ADC includes single-use bioreactors and purification suites with equipment for Good Manufacturing Practice (GMP) production of vaccines and biologics for clinical trials, including the capability of producing sterile vaccines in glass vials.
The ADC is an approximately 45,000 square foot BSL-2 facility which can employ up to 70 researchers, scientists, manufacturing, and technical support staff. This facility was decommissioned in May 2024, and is ready to be reactivated on the earlier of 2027 or in the case of a national or international emergency.
Commercialization
Following FDA approval, we initiated the commercial launch of TONMYA in 2025, our first internally developed and commercialized product. We have established a focused commercial infrastructure and team designed to support launch execution, drive prescriber awareness, and enable patient access, while maintaining compliance with applicable regulatory and industry standards.
We commercialize TONMYA through our sales organization, which includes an internal sales force, and a contracted salesforce, non-personal promotion, digital engagement, market access programs, patient support services, and distribution through national wholesalers and specialty distributors. We are focused on obtaining payer coverage, building physician awareness, and driving adoption primarily in practices that have a history of diagnosing and treating fibromyalgia, which includes rheumatology, primary care, pain management and neurology practices.
We have contracted for two commercial supply sources of TONMYA in the U.S., one of which is Almac Pharma Services, a member of the privately owned Almac Group.
We have managed the continued commercial operations for Zembrace Symtouch and Tosymra, following the acquisition of these products in 2023.
Marketing, Sales and Distribution
Marketing activity for TONMYA, Zembrace Symtouch and Tosymra in the United States is conducted by our wholly-owned subsidiary, Tonix Medicines, Inc. We focus our sales and marketing efforts on physicians in private practice and in public treatment systems. We employ standard pharmaceutical marketing practices to promote our products, encompassing advertisements, professional symposia, sales initiatives, and educational outreach aimed at physicians, nurses, social workers, counselors, and other stakeholders involved in treating fibromyalgia and acute migraine in adults. We have established contracts with third-party vendors to handle logistics, offer customer services, and manage other related aspects for our products. These services include managing product-specific websites, conducting insurance research, processing orders, and handling delivery and fulfillment services. TONMYA, Zembrace Symtouch and Tosymra are primarily sold to pharmaceutical wholesalers, pharmacies, and specialty distributors. We have implemented patient access programs and expand distribution channels in our marketing efforts for our fibromyalgia and migraine drugs.
20
In 2024, Tonix engaged EVERSANA to support the launch strategy and commercial planning of TONMYA for the treatment of fibromyalgia. Specifically, EVERSANA also worked with Tonix to assess the fibromyalgia landscape and help plan an efficient go-to-market strategy.
Our commercialization strategy for TONMYA is centered on a cost-efficient, targeted sales and marketing approach focused on the top prescribing healthcare providers who treat patients with fibromyalgia, including primary care physicians and relevant specialists. Our field-based promotion is conducted through a combination of internal commercial personnel and contracted sales representatives. As of December 31, 2025, our commercial organization included approximately 90 field sales representatives, inclusive of internal and contracted resources, supported by sales leadership, training, and operations functions.
In addition to field-based promotion, we utilize non-personal promotion and digital marketing initiatives to support disease awareness, product education, and appropriate utilization. These efforts are intended to complement in-person engagement and expand reach within our target prescriber universe. We have 11 employees supporting sales administration and marketing initiatives; customer service requests and top-tier headache specialists. We utilize third party vendors to support trade, managed markets, marketing initiatives, and promotional compliance programs.
TONMYA is distributed in the United States through national pharmaceutical wholesalers and specialty distributors pursuant to customary commercial arrangements. Tonix has contracted with its existing wholesalers and specialty pharmacies for the distribution of TONMYA. We utilize third-party logistics providers to support product warehousing, order fulfillment, and distribution activities. Our distribution and supply chain operations are structured to comply with applicable requirements, including those under the Drug Supply Chain Security Act (DSCSA). Product returns are managed in accordance with industry-standard practices.
Market Access and Patient Support
We have implemented a market access strategy intended to support broad and appropriate patient access to TONMYA across key payer segments, including commercial plans and government programs. Our market access efforts include engagement with payers and pharmacy benefit managers, formulary review processes, and contracting activities customary for prescription pharmaceutical products.
We also offer patient support services designed to assist with benefits verification, prior authorization support, and access to available patient assistance programs. These services are administered through third-party vendors and are intended to facilitate appropriate initiation and continuation of therapy, consistent with applicable laws and regulations.
Commercial Compliance and Oversight
We maintain policies, procedures, and controls designed to support compliant commercial operations, including a promotional review committee (PRC) process for review and approval of promotional and non-promotional materials, field force compliance training, and pharmacovigilance and adverse event reporting procedures. These activities are intended to support compliant engagement with healthcare professionals, patients, and other stakeholders as we execute our commercial strategy.
Competition
Our sector faces intense competition and experiences rapid, substantial technological advancements both domestically and internationally. Our potential competitors encompass major pharmaceutical and biotechnology firms, specialty pharmaceutical and generic drug manufacturers, academic institutions, government agencies, and research organizations. We consider efficacy, safety, tolerability, reliability, pricing, and reimbursement levels as crucial competitive factors influencing the development and commercial success of our product candidates. Numerous potential competitors, including some of the organizations listed below, possess considerably larger financial, technical, and human resources, as well as extensive experience in discovering and developing product candidates, securing FDA and other regulatory approvals, and commercializing those products, far surpassing our own capabilities. Hence, our competitors might achieve greater success in securing FDA approval for drugs and gaining widespread market acceptance compared to us. The drugs offered by our competitors may prove to be more effective or better marketed and sold than any product we bring to market, potentially rendering our product candidates obsolete or non-competitive before we can recoup the expenses incurred in their development and commercialization. We expect to encounter heightened competition as the market sees the introduction of new drugs and the emergence of advanced technologies. Additionally, the evolution of novel treatment approaches for the conditions we are focusing on may potentially diminish the competitiveness or relevance of our drugs. Below, we provide an overview of the competitive landscape for the indications where Tonix has product candidates either in or nearing the clinical stages of development.
21
Fibromyalgia
As of 2026, the U.S. fibromyalgia therapeutic landscape consists of four FDA-approved treatments.. The first three products - pregabalin (Lyrica ®), duloxetine (Cymbalta a®), and milnacipran (Savella a®) - received FDA approval between 2007 and 2009 and have represented the standard of care for more than a decade. In August 2025, FDA approved TONMYA, the first new fibromyalgia treatment in more than 15 years and the first approved therapy aimed at improving non-restorative sleep, a core contributor to fibromyalgia pain and fatigue. The U.S. fibromyalgia market has historically been characterized by limited innovation and treatment dissatisfaction, with many patients cycling through therapies or relying on off-label analgesics due to incomplete symptom control. The approval of TONMYA introduces a differentiated mechanism targeting nonrestorative sleep.
We actively monitor multiple companies advancing investigational therapies for fibromyalgia. In the U.S., Axsome Therapeutics, Inc. initiated their Phase 3 trial of AXS-14 (esreboxetine) for the management of fibromyalgia in January 2026. Dogwood Therapeutics, Inc. (formerly Virios Therapeutics, Inc.) continues the development of IMC-1, a fixed-dose combination of famciclovir + celecoxib. Outside the U.S., Ono Pharmaceutical is advancing ONO-1110 as an oral medication designed for endocannabinoid regulation. The drug is being developed in Japan for several indications, including fibromyalgia, postherpetic neuralgia, and major depressive disorder. Tryptamine Therapeutics Ltd. reported positive Phase IIa data for TRP-8802 in fibromyalgia at the 2024 IASP meeting.
Migraine
Zembrace SymTouch and Tosymra continue to serve the acute migraine market as branded formulations of sumatriptan, competing directly with generic subcutaneous and intranasal products. Beyond generic pressure, both brands now operate within an increasingly competitive landscape shaped by the growth of next-generation acute therapies, including oral CGRP antagonists such as Pfizer’s Nurtec® ODT (rimegepant) and AbbVie’s Ubrelvy® (ubrogepant) and Qulipta® (atogepant), as well as intranasal CGRP options like Pfizer’s Zavzpret™ (zavegepant).
The competitive environment expanded further in 2025 with the FDA approval of Axsome Therapeutic Inc.’s Symbravo® (meloxicam and rizatriptan). Satsuma Pharmaceuticals, Inc. received FDA approval for Atzumi, a nasal powder formulation of DHE (dihydroergotamine) for acute treatment of migraine.
Meanwhile, broader migraine development pipelines including but not limited to assets such as PUR-3100, an orally inhaled dry-powder formulation of dihydroergotamine (DHE) developed by Pulmatrix, Inc., signal ongoing innovation and expected market expansion through 2026 as companies advance differentiated mechanisms and delivery formats.
Major Depressive Disorder
Many antidepressant medications are beyond their patent life and are generally produced by generic drug companies, including several compounds in the tricyclic class (e.g., amitriptyline), the serotonin-selective reuptake inhibitor class (e.g., fluoxetine, paroxetine and sertraline), the serotonin-norepinephrine reuptake inhibitor class (e.g., venlafaxine, duloxetine), as well as the norepinephrine-dopamine reuptake inhibitor, bupropion. Tonix is aware of several companies developing novel prescription medicines for depression, including companies with late-stage (Phase 3) clinical-stage programs, including but not limited to: Johnson & Johnson, Inc., Luye Pharma Group, Ltd., Definium Therapeutics, Inc., and Vanda Pharmaceuticals, Inc.
Acute Stress Reaction/Acute Stress Disorder (ASR/ASD)
There are currently no approved drugs specifically for treating acute stress reaction (ASR) or for the prevention of acute stress disorder (ASD). As of late 2025, BXCL501 (dexmedetomidine sublingual film) which is the FDA-approved drug marketed as IGALMI® remains under investigation for stress-related indications but is not approved for ASR, ASD, or PTSD. BioXcel Therapeutics, with U.S. Department of Defense support, expects to begin a Phase 2a trial of BXCL501 in patients experiencing ASD following motor vehicle collisions.
22
Lyme Disease
There are no approved drugs for Lyme disease prevention. We are aware of several companies currently developing Lyme Disease vaccines. Pfizer, in collaboration with Valneva, is developing VLA15, a multivalent recombinant protein subunit vaccine designed to prevent Lyme in the U.S. and Europe. The program has completed its Phase 3 VALOR trial vaccinations, with a data readout expected in the first half of 2026.
Moderna is advancing two mRNA-based vaccine candidates as part of its bacterial vaccine program (mRNA-1982 and mRNA-1975), which are in Phase I/II clinical trials.
Transplantation Rejection and Autoimmune Treatments with Anti-CD40-ligand Monoclonal Antibodies
We are aware of multiple companies advancing the development of biologics targeting the CD40L molecule. Sanofi’s Frexalimab (SAR441344) is in Phase III development for Multiple Sclerosis. Frexalimab is also being advanced in clinical trials for other autoimmune diseases, including Systemic Lupus Erythematosus. Eledon Pharmaceuticals is advancing tegoprubart for organ transplant rejection. The company completed its Phase II (BESTOW) trial in 2025 and plans to meet with the FDA to discuss Phase III trial design and data requirements in 2026. Amgen Inc. is currently conducting a Phase III trial to test dazodalibep for Sjogren’s Syndrome, with an estimated completion date for the second half of 2026. In November 2024, UCB and Biogen announced the initiation of their potentially confirmatory Phase III trial to test dapirolizumab Pegol for Systemic Lupus Erythematosus. Biogen expects topline data readouts in late 2027 or early 2028.
These are a few key highlights that underscore the focus of major pharmaceutical companies in this area and we monitor the activity of other companies in the process of developing antagonistic anti-CD40 mAbs, including Novartis, Boehringer Ingelheim GmbH, Kiniska Pharmaceuticals, Boston Immune Therapies, and NapaJen Pharma Inc.
Prader Willi Syndrome
Vykat® (Diazoxide Choline), developed by Soleno Therapeutics, is approved for the treatment of Prader Willi Syndrome (PWS). Tonix is aware of several companies currently developing treatments for PWS, including Harmony Biosciences and Aardvark Therapeutics. Harmony Biosciences is advancing pitolisant in a Phase III clinical trial (TEMPO).
Gastric and Colorectal Cancer
We are aware of several companies that are focused on developing treatments for Gastric and Colorectal Cancer, including but not limited to: Johnson & Johnson, AbbVie Inc., AstraZeneca PLC, Roche Holding AG, Novartis AG, Merck & Co. Inc., Pfizer Inc., GSL plc, Bristol Myers Squibb Company, BioNtech SE and Jazz Pharmaceuticals.
Smallpox / Mpox
There are multiple approved vaccines globally for the prevention of smallpox and mpox including Jynneos®, developed by Bavarian Nordic, ACAM2000®, developed by Emergent BioSolutions, and LC16m8®, developed by KM Biologics. We are aware of several companies currently developing next-generation vaccines for smallpox and mpox, including but not limited to: Moderna, BioNTech SE, GeoVax and NonoViricides.
Cocaine Intoxication
There are no approved antidotes for the treatment of cocaine intoxication. Patients generally receive supportive care. We are not aware of any drugs in development for the treatment of cocaine intoxication.
Intellectual Property
We believe that we have an extensive patent portfolio and substantial know-how relating to TNX-102 SL, Zembrace®, Tosymra®, TNX-1300, TNX-1500, TNX-2900, TNX-1900, TNX-801, TNX-1800 and TNX-1700, and our other product candidates. Our patent portfolio, described more fully below, includes claims directed to various compositions and methods of use related to our product candidates. As of March 5, 2026, the patents we are either the owner of record of or own the contractual right to include 45 issued U.S. patents and 46 issued non-U.S. patents. We are actively pursuing an additional 22 U.S. non-provisional patent applications, 4 international patent applications (PCT), and 238 non-U.S./non-PCT patent applications.
23
We strive to protect the proprietary technology that we believe is important to our business, including our proprietary technology platform, our product candidates, and our processes. We seek patent protection in the U.S. and internationally for our products, their methods of use and processes of manufacture, and any other technology to which we have rights, where available and when appropriate. We also rely on trade secrets that may be important to the development of our business.
Our success will depend on 1) the ability to obtain and maintain patent and other proprietary rights in commercially important technology, inventions and know-how related to our business, 2) the validity and enforceability of our patents, 3) the continued confidentiality of our trade secrets, and 4) our ability to operate without infringing the valid and enforceable patents and proprietary rights of third parties. We also rely on continuing technological innovation and in-licensing opportunities to develop and maintain our proprietary position.
We cannot be certain that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications we may own or license in the future, nor can we be certain that any of our existing patents or any patents we may own or license in the future will be useful in protecting our technology. For this and more comprehensive risks related to our intellectual property, please see “Risk Factors — Risks Relating to Our Intellectual Property.”
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the date of filing the first non-provisional priority application. In the United States, a patent’s term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the PTO in granting a patent or may be shortened if a patent is terminally disclaimed over another patent.
The term of a U.S. patent that covers a drug approved by the FDA or methods of making or using that drug may also be eligible for patent term extension, which permits patent term restoration as compensation for the patent term lost during the FDA regulatory review process. The Drug Price Competition and Patent Term Restoration Act, also known as the Hatch-Waxman Act, is a federal law that encourages new drug research by restoring patent term lost to regulatory delays by permitting a patent term extension of up to five years beyond the statutory 20-year term of the patent for the approved product or its methods of manufacture or use if the active ingredient has not been previously approved in the U.S. The length of the patent term extension is related to the length of time the drug is under regulatory review. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only one patent applicable to an approved drug may be extended. Similar provisions are available in Europe and some other foreign jurisdictions to extend the term of a patent that covers an approved drug.
When possible, depending upon the length of clinical trials and other factors involved in the filing of an NDA, we expect to apply for patent term extensions for patents covering our product candidates and their methods of manufacture or use.
The patent portfolios for our proprietary technology platform and our most advanced product candidates as of March 5, 2026 are summarized below.
TNX-102 SL — Central Nervous System Conditions
Our patent portfolio for TNX-102 SL includes patents and patent applications directed to compositions of matter of CBP, formulations containing CBP, and methods for treating CNS conditions, such as TNX-102 SL for PTSD, for pain, fatigue and sleep disturbances in fibromyalgia, for treating or managing fibromyalgia (early onset response, favorable tolerability, or side effect profile), for alcohol abuse, for disordered sleep, for sexual dysfunction, for depression in fibromyalgia, for fatigue and disordered sleep (e.g., CAP rates), for post-acute sequelae of SARS-CoV-2 infection, for acute stress reaction/acute stress disorder, and for agitation in neurodegenerative conditions, e.g., AD.
Certain eutectic compositions were discovered by development partners and are termed the “Eutectic Technology.” The patent portfolio for CBP compositions (e.g., TNX-102 SL) relating to the Eutectic Technology includes patents and patent applications directed to eutectic compositions containing CBP, eutectic CBP formulations, methods for treating PTSD and other CNS conditions utilizing eutectic CBP compositions and formulations, and methods of manufacturing eutectic CBP compositions. The Eutectic Technology patent portfolio includes U.S. patents, such as U.S. Patent No. 9,636,408, U.S. Patent No. 9,956,188, U.S. Patent No. 10,117,936, U.S. Patent No. 10,357,465, U.S. Patent No. 10,864,175, U.S. Patent No. 11,026,898, and U.S. Patent No. 11,839,594. These U.S. patents and counterpart non-U.S. patents, and any U.S. and non-U.S. patents that issue in the future from this portfolio would expire in 2034 or 2035, excluding any patent term adjustments or extensions.
The unique pharmacokinetic profile of TNX-102 SL, or the PK Technology, was discovered by Tonix and its development partners. The patent portfolio for TNX-102 SL relating to the PK Technology includes patent applications directed to compositions of matter of CBP, formulations containing CBP, methods for treating PTSD, agitation in neurodegenerative conditions, and other CNS conditions utilizing these compositions and formulations. The PK Technology patent portfolio includes U.S. Patent Application No. 19/409,704. If U.S. and non-U.S. patents claiming priority from those applications issue, those patents would expire in 2033, excluding any patent term adjustments or extensions.
24
On May 2, 2017, U.S. Patent No. 9,636,408 entitled “Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride”, issued. The patent claims recite pharmaceutical compositions comprising the eutectic. The patent claims also recite methods of manufacturing the eutectic.
On September 13, 2017, European patent 2,501,234, entitled “Methods and Compositions for Treating Symptoms Associated with PTSD Using Cyclobenzaprine”, issued. This patent recites the use of CBP for the treatment of PTSD. On January 11, 2024, the European Patent Office Technical Board of Appeal reversed the October 2019 decision of the Opposition Division of the European Patent Office maintaining the patent in unamended form and held the patent to be invalid. No appeal may be taken from that decision.
On December 15, 2017, Japanese Patent No. 6259452, entitled “Compositions and Methods for Transmucosal Absorption,” issued. These claims relate to the pharmacokinetic profile of TNX-102 SL.
On August 3, 2022, European Patent No. 2861223, entitled “Compositions and Methods for Transmucosal Absorption,” issued. These claims relate to the pharmacokinetic profile of TNX-102 SL.
On March 20, 2018, U.S. Patent No. 9,918,948 entitled “Methods and Compositions for Treating Symptoms Associated with PTSD Using Cyclobenzaprine,” issued. The claims recite a method of using TNX-102 SL’s active ingredient cyclobenzaprine to treat PTSD and provides US market exclusivity until 2030, excluding any patent term extensions.
On March 23, 2018, Japanese Patent No. 6310542 entitled “Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride”, issued. The claims recite pharmaceutical compositions comprising the eutectics and methods of manufacturing these eutectic formulations.
On May 1, 2018, U.S. Patent No. 9,956,188, entitled “Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride”, issued. The claims recite a eutectic of cyclobenzaprine hydrochloride and mannitol and methods of making those eutectics.
On November 6, 2018, U.S. Patent No. 10,117,936, entitled “Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride”, issued. The claims recite pharmaceutical compositions of eutectics of cyclobenzaprine hydrochloride and mannitol and methods of making those compositions.
On April 16, 2019, Chinese Patent No. ZL 201480024011.1 entitled “Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride”, issued. The claims recite pharmaceutical compositions comprising eutectics of cyclobenzaprine hydrochloride and mannitol and methods of making those compositions.
On August 4, 2023, Chinese Patent No. ZL201910263541.6, entitled “Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride,” issued. The claims recite a eutectics of cyclobenzaprine hydrochloride and beta-mannitol and methods of making those eutectics.
On July 23, 2019, U.S. Patent No. 10,357,465 entitled “Eutectic Formulations of Cyclobenzaprine Hydrochloride”, issued. The claims recite a eutectic of cyclobenzaprine hydrochloride and mannitol and methods of making those eutectics.
On December 11, 2019, European patent 2968992, entitled “Eutectic Formulations of Cyclobenzaprine Hydrochloride”, issued. This patent recites pharmaceutical compositions comprising a eutectic of mannitol and Cyclobenzaprine HCl and methods of making the same. In response to an opposition filed in September 2020 by Hexal AG, the European Patent Office’s Opposition Division upheld the patent in unamended form after the January 2022 oral proceedings. Hexal AG did not appeal that decision.
On December 25, 2019, European patent 2,683,245, entitled “Methods and Compositions for Treating Depression Using Cyclobenzaprine”, issued. The claims recite the use of CBP for the treatment of depression in a FM patient. This patent provides TNX-102 SL with European market exclusivity until March 2032 and may be extended based on the timing of the European marketing authorization of TNX-102 SL for depression in a FM patient. In September 2020, Hexal AG filed an opposition against this patent. The European Patent Office’s Opposition Division upheld the patent claims in unamended form after the February 2022 oral proceedings. Hexal AG did not appeal that decision.
25
On June 4, 2024, U.S. Patent No. 11,998,516, entitled “Methods and Compositions for Treating Depression Using Cyclobenzaprine,” issued. The claims recite methods for treating major depressive disorder in a fibromyalgia patient using a composition comprising cyclobenzaprine or its salts.
On December 15, 2020, U.S. Patent No. 10,864,175 entitled “Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride”, issued. The claims recite a eutectic comprising cyclobenzaprine hydrochloride and beta-mannitol.
On December 12, 2023, U.S. Patent No. 11,839,594 entitled “Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride”, issued. The claims recite a method of manufacturing a eutectic comprising cyclobenzaprine hydrochloride and beta-mannitol comprising mixing or milling.
On February 14, 2024, European Patent No. 3,650,081, entitled “Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride”, issued. The claims recite a eutectic of mannitol and cyclobenzaprine hydrochloride and methods of manufacturing a eutectic.
On April 8, 2021, U.S. non-provisional Patent Application No. 17/226,058 and International Patent Application No. PCT/US2021/026492, entitled “Cyclobenzaprine Treatment for Sexual Dysfunction” were filed. The PCT application is now nationalized in Australia, Canada, China, European Patent Office, Japan, and Hong Kong. On October 5, 2022, International Patent Application No. PCT/US2022/045791, entitled “Cyclobenzaprine Treatment for Sexual Dysfunction,” was filed and is now nationalized in European Patent Office and U.S. (U.S. Patent Application No. 18/698,483). The claims of these applications are directed to methods using pharmaceutical compositions and combinations for treating sexual dysfunction with cyclobenzaprine or pharmaceutically acceptable salts of cyclobenzaprine.
On October 25, 2016 and July 28, 2020, U.S. Patent No. 9,474,728 and U.S. Patent No. 10,722,478, entitled “Methods and Compositions for Treating Fatigue Associated with Disordered Sleep Using Very Low Dose Cyclobenzaprine”, issued, respectively. The claims are directed to a method for monitoring the effectiveness of cyclobenzaprine treatment for disordered sleep and method for reducing CAP rates A2 or A3 by treating a subject with a pharmaceutical composition comprising cyclobenzaprine.
On December 11, 2018, International Patent Application No. PCT/IB2018/001509, entitled “Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions,” was filed. The PCT application is now nationalized in 16 countries. The claims are directed to methods for treating or preventing agitation, cognitive decline, psychosis, and associated symptoms thereof using pharmaceutical compositions and combinations with cyclobenzaprine or pharmaceutically acceptable salts of cyclobenzaprine.
On November 28, 2023, U.S. Patent No. 11,826,321, entitled “Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions,” issued. The claims are directed to a method for treating or preventing one or more agitation associated symptoms comprising administering a eutectic of cyclobenzaprine HCl and mannitol.
On August 20, 2019, International Patent Application No. PCT/IB2019/000940, entitled “Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder,” was filed. The PCT application is now nationalized in 18 countries. The claims are directed to methods of treating acute stress disorder or post-traumatic stress disorder in a subject who has experienced a traumatic event using pharmaceutical compositions with cyclobenzaprine, amitriptyline or pharmaceutically acceptable salts of cyclobenzaprine or amitriptyline.
On November 19, 2021, International Patent Application No. PCT/US2021/060011, entitled “Cyclobenzaprine Treatment for Alcohol Use Disorder,” was filed. The PCT application is now nationalized in 13 countries. The claims are directed to methods for treating alcohol use disorder and associated symptoms using pharmaceutical compositions with cyclobenzaprine or pharmaceutically acceptable salts of cyclobenzaprine.
On December 7, 2021, International Patent Application No. PCT/US2021/062244, entitled, “Cyclobenzaprine Treatment for Fibromyalgia,” was filed. The PCT is now nationalized in 15 countries. The claims are directed to methods for treating fibromyalgia and its associated symptoms of pain, sleep disturbance and/or fatigue by transmucosally administering a eutectic of cyclobenzaprine hydrochloride and mannitol in dosage units with a basifying agent.
On June 21, 2023, U.S. non-provisional Patent Application No. 18/212,500 and International Patent Application No. PCT/US2023/025895, entitled “Cyclobenzaprine Treatment for Post-Acute Sequelae of (SARS)-CoV-2 Infection (PASC),” were filed. The PCT is now nationalized in 9 countries. The claims are directed to methods of treating PASC or one or more associated symptoms comprising administering cyclobenzaprine or a pharmaceutically acceptable salts of cyclobenzaprine.
26
On December 19, 2024, U.S. non-provisional Patent Application No. 18/988,194 and International Patent Application No. PCT/US2024/061125, entitled “Early Onset Response, Favorable Tolerability, and Side Effect Profile in the Treatment of Fibromyalgia,” were filed. The claims are directed to methods for treating or managing fibromyalgia and its associated symptoms in subjects characterized by an early onset of one or more of: (1) a reduction in widespread pain; (2) a reduction in sleep disturbance; (3) a reduction in fatigue; or (4) an improved sleep quality. The claims are also directed to methods for preventing or avoiding clinically meaningful changes in mean weight, in mean systolic blood pressure or mean diastolic blood pressure, or a decline in sexual functioning.
On January 23, 2025, International Patent Application No. PCT/US2025/012803, entitled Cyclobenzaprine Treatment for Acute Stress Reaction or Acute Stress Disorder,” was filed. The claims are directed to methods for treating or preventing acute stress reaction (ASR) or acute stress disorder (ASD) and associated symptoms thereof using a composition with cyclobenzaprine or its salts in.
TNX-1900 — Oxytocin-Based Treatments for the Treatment of Binge Eating Disorder, Adolescent Obesity, Bone Health in Pediatric Autism, and Arginine-Vasopressin Deficiency
We have acquired the migraine and pain treatment technologies of Trigemina, Inc., and have assumed its license rights to related technologies from The Board of Trustees of the Leland Stanford Junior University. TNX-1900, an enhanced formulation of nasal oxytocin, has demonstrated activity in several non-clinical studies in pain, including migraine.
As part of our acquisition, we acquired International Patent Application No. PCT/US2016/012512, filed on January 7, 2016, entitled “Magnesium-Containing Oxytocin Formulations and Methods of Use” (nationalized in 13 countries). We also acquired U.S. Patent Nos. 9,629,894 and 11,389,473, entitled “Magnesium-Containing Oxytocin Formulations and Methods of Use”, which will expire in January 2036, excluding any patent term extensions. On July 1, 2022, Chinese Patent No. ZL201680013809.5, entitled “Magnesium-Containing Oxytocin Formulations and Methods of Use,” issued. On October 4, 2023, European Patent No. 3242676, entitled “Magnesium-Containing Oxytocin Formulations and Methods of Use,” issued.
We also acquired International Patent Application No. PCT/US2017/027265, filed April 12, 2017, entitled “Magnesium-Containing Oxytocin Formulations and Methods of Use” (nationalized in 9 countries). On December 3, 2024, U.S. Patent No. 12,156,897, entitled “Magnesium-Containing Oxytocin Formulations and Methods of Use,” issued. The patent, which claims methods for treating autism spectrum disorder, social anxiety disorder, or a social communication disorder, will expire in April 2037, excluding any patent term extensions. On January 1, 2023, Chinese Patent No. ZL201780036185.3, entitled “Magnesium-Containing Oxytocin Formulations and Methods of Use,” issued. On November 13, 2025 Japan Patent No. 7093559, entitled “Magnesium-Containing Oxytocin Formulations and Methods of Use,” issued.
We also have rights to International Patent Application No. PCT/US2019/020419, filed on April 12, 2017, entitled “Labeled Oxytocin and Method of Manufacture and Use” (nationalized in the U.S., European Patent Office and Japan). On April 30, 2024, U.S. Patent No. 11,970,554, entitled “Labeled Oxytocin and Method of Manufacture and Use,” issued. On November 11, 2025, U.S. Patent No. 12,466,855, entitled “Labeled Oxytocin and Method of Manufacture and Use,” issued.
We have entered into an exclusive license to the University of Geneva’s technology for using oxytocin to treat insulin resistance and related syndromes, including obesity. This license expands our intranasal potentiated oxytocin development program, TNX-1900, into cardiometabolic syndromes. Under the license, we have rights to European Patent No. EP2571511B1, entitled “New Uses of Oxytocin-like Molecules and Related Methods.” We also have rights to U.S. Patent No. 9,101,569, entitled “Methods for the Treatment of Insulin Resistance.” The U.S. and non-U.S. patents expire in May 2031, excluding any patent term adjustments or extensions.
We are developing oxytocin peptide analogs for headache disorders. On March 18, 2025, we filed International Patent Application No. PCT/US2025/020483, entitled “Oxytocin Peptide Analogs.”
TNX-2900 — Oxytocin-Based Therapeutics Treatments for Prader-Willi Syndrome (PWS)
We have licensed technology using oxytocin-based therapeutics for the treatment of PWS and non-organic failure to thrive disease from the French National Institute of Health and Medical Research (INSERM). The co-exclusive license relates to TNX-2900, an intranasal potentiated oxytocin, for the treatment of Prader-Willi syndrome and other feeding disorders. Under the license, we have rights to European Patent No. EP2575853B1, entitled “Methods and Pharmaceutical Composition for the Treatment of a Feeding Disorder with Early-Onset in a Patient”; U.S. Patent No. 8,853,158, entitled “Methods for the Treatment of a Feeding Disorder with Onset During Neonate Development Using an Agonist of the Oxytocin Receptor”; and U.S. Patent No. 9,125,862, entitled “Methods for the Treatment of Prader-Willi-like Syndrome or Non-Organic Failure to Thrive (NOFITT) Feeding Disorder Using an Agonist of the Oxytocin Receptor.” The U.S. and non-U.S. patents expire in May 2031, excluding any patent term extensions.
27
TNX-1300 — Cocaine Intoxication Treatment
We have licensed rights from The Trustees of Columbia University in the City of New York, The Regents of the University of Michigan, and University of Kentucky Research Foundation to develop a potential product, TNX-1300, for the treatment of cocaine intoxication. The licensed patents are directed to mutant cocaine esterase polypeptides and methods of using these polypeptides as anti-cocaine therapeutics. They include U.S. Patent Nos. 8,318,156 and 9,200,265, entitled “Anti-Cocaine Compositions and Treatment” and various counterpart patents outside of the U.S (e.g., European Patent 2046368). These patents provide TNX-1300 with US market exclusivity until February 2029, and market exclusivity outside of the U.S. until July 10, 2027, subject to any patent term extensions.
TNX-1500 — anti-CD40L Therapeutics
We are developing TNX-1500, a humanized mAb that targets CD40L for the prevention and treatment of organ transplant rejection. In this regard, we filed International Application No. PCT/EP2020/068589, entitled “Anti-CD154 antibodies and uses thereof” on July 1, 2020 (nationalized in 15 countries). We also filed International Patent Application No. PCT/US2020/028002 on April 13, 2020, entitled “Inhibitors of CD40-CD154 Binding” (nationalized in U.S., Canada, China, European Patent Office and Japan). We also filed International Patent Application No. PCT/US2022/011404, entitled “Methods of Inducing Immune Tolerance with Modified Anti-CD154 Antibodies” on January 6, 2022 (nationalized in 14 countries). We also filed International Patent Application No. PCT/EP2025/065085, entitled “Treatment Methods Comprising Administration of Modified CD154 Antibodies” on May 30, 2025.
On August 28, 2024, European Patent No. 3993876, entitled “Anti-CD154 Antibodies and Uses Thereof,” issued (validated in 37 countries). The claims recite an isolated antibody that binds CD154, compositions comprising the antibody, and use of the compositions for treating or preventing a transplant rejection.
On April 22, 2025, Chinese Patent No. ZL202080059891.1, entitled “Anti-CD154 antibodies and uses thereof,” issued. The claims recite an isolated antibody that binds CD154, compositions comprising the antibody, and uses of the antibody for inhibiting an immune response, treating or preventing a transplant rejection, or inhibiting xenotransplant rejection.
On November 14, 2025, Japanese Patent No. 7775080, entitled “Anti-CD154 antibodies and uses thereof,” issued. The claims recite an antibody or isolated antibody that binds CD154 and compositions comprising the antibody for use in treating or preventing a transplant rejection, inducing hematopoietic chimerism in a transplant recipient, inducing central tolerance in a transplant recipient, or inhibiting xenotransplant rejection.
TNX-801 — Live Horsepox Vaccine for Prevention of Smallpox and Mpox
We own the rights to develop a potential biodefense technology, TNX-801, a live horsepox that is being developed as a new smallpox and mpox preventing vaccine, we have filed patent applications directed to synthetic chimeric poxviruses and methods of using these poxviruses to protect individuals against smallpox. These applications include U.S. non-provisional Patent Application No. 15/802,189 and International Patent Application No. PCT/US2017/059782 (nationalized in 15 countries and filed in 4 non-PCT countries). We also own the rights to develop other vaccine candidates against smallpox. With respect to these vaccine candidates, we own International Patent Application No. PCT/US2019/030486 and the non-convention and national phase applications related thereto (nationalized in 17 countries and filed in 2 non-PCT countries). The smallpox vaccine technologies relate to proprietary forms of live horsepox and vaccinia vaccines which may be safer than ACAM2000, the only currently available replication competent, live vaccinia vaccine to protect against smallpox disease. We believe that this technology, after further development, may be of interest to biodefense agencies in the U.S. and other countries.
On May 31, 2022, U.S. Patent No. 11,345,896 was issued. The claims recite a synthetic chimeric orthopoxvirus (scOPV), a synthetic chimeric horsepox virus (scHPXV), methods of generating the scOPV and scHPXV, and compositions comprising the scOPV or scHPXV.
On July 22, 2025, U.S. Patent No. 12,365,879, entitled “Synthetic Chimeric Poxviruses,” issued. The claims recite methods of producing a synthetic chimeric orthopoxvirus (scOPV) and a synthetic chimeric horsepox virus (scHPXV) and methods of treating infections using the scOPV or scHPXV.
On April 2, 2024, Chinese Patent No. ZL201780078546.0, entitled “Synthetic Chimeric Poxviruses,” issued. The claims recite a synthetic chimeric orthopoxvirus (scOPV), methods of producing an scOPV, compositions comprising the scOPV, and uses of the scOPV.
TNX-1800 and TNX-1850 — Live Modified Horsepox Vaccine for Prevention of COVID
We are developing TNX-1800 and TNX-1850, live minimally replicative modified HPXVs, as a COVID preventing vaccine against different strains of SARS-CoV-2. On February 26, 2021, we filed International Patent Application No. PCT/US2021/020119, entitled “Recombinant Poxvirus Based Vaccine Against SARS-CoV-2.” On the same date, we also filed applications in Argentina and Taiwan and we filed U.S. Application No. 17/187,678. The PCT application is now nationalized in 19 countries. These applications are directed to synthetic poxviruses comprising a SARS-CoV-2 virus protein, poxvirus delivery vectors for SARS-CoV-2 virus proteins and methods of using these modified poxviruses to protect individuals against COVID.
28
TNX-1700 — Recombinant Trefoil Family Factor 2 (rTFF2) to Treat Gastric and Colorectal Cancers
We have licensed rights from The Trustees of Columbia University in the City of New York to develop a potential product, TNX-1700, for the treatment of gastric and colorectal cancers. The licensed patents are directed to rTFF2 compositions and methods of treatment. The licensed patents, U.S. Patent No. 10,124,037 and U.S. Patent No. 11,167,010, provide TNX-1700 with US market exclusivity until April 2033, subject to any patent term extensions. On August 27, 2020, we filed International Patent Application No. PCT/IB2020/000699 entitled “Modified TFF2 Polypeptides.” The PCT application is now nationalized in 12 countries.
Zembrace and Tosymra — Sumatriptan
We have acquired the intellectual property rights of Zembrace SymTouch and Tosymra and their uses in treating migraine from Upsher-Smith Laboratories, LLC. These rights include U.S. Patent No. 9,211, 282, U.S. Patent No. 9,610,280, U.S. Patent No. 9,974,770, U.S. Patent No. 10,603,305, U.S. Patent No. 11,337,962, U.S. Patent No. 10,537,554, and U.S. Patent No. 11,364, 224. These rights also include International Patent Application No. PCT/US2016/015961, entitled “Pharmaceutical Composition Comprising Sumatriptan for Treating Migraine,” (nationalized in 7 countries, excluding rights in Brazil and China) and International Patent Application No. PCT/IB2010/001708, entitled “Formulations Comprising Triptan Compounds,” (nationalized in 11 countries, excluding rights in Brazil, Russia, India, and China).
Trade Secrets
In addition to patents, we rely on trade secrets and know-how to develop and maintain our competitive position. For example, significant aspects of our proprietary technology platform are based on unpatented trade secrets and know-how. Trade secrets and know-how can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by confidentiality agreements and invention assignment agreements with our employees, consultants, scientific advisors, contractors, and commercial partners. These agreements are designed to protect our proprietary information and, in the case of the invention assignment agreements, to grant us ownership of technologies that are developed through a relationship with a third party. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, agreements or security measures may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our contractors use intellectual property owned by others in their work for us, disputes may arise as to rights in related or resulting inventions and know-how.
29
Issued Patents
Our current patents owned or licensed include:
Anti-Cocaine Therapeutics
| Patent No. | Title | Country / Region | Expiration Date | |||
| 8,318,156 | Anti-Cocaine Compositions and Treatment | U.S.A. | February 14, 2029 | |||
| 9,200,265 | Anti-Cocaine Compositions and Treatment | U.S.A. | December 30, 2027 | |||
| 2007272955 | Anti-Cocaine Compositions and Treatment | Australia | July 10, 2027 | |||
| 2014201653 | Anti-Cocaine Compositions and Treatment | Australia | July 10, 2027 | |||
| 2657246 | Anti-Cocaine Compositions and Treatment | Canada | July 10, 2027 | |||
| 612929 | Anti-Cocaine Compositions and Treatment | New Zealand | July 10, 2027 | |||
| 2046368 (602007045044.6 in Germany; 502016000056543 in Italy) | Anti-Cocaine Compositions and Treatment | European Patent Office – Germany, Spain, France, United Kingdom, and Italy | July 10, 2027 | |||
| 2009/00197 | Anti-Cocaine Compositions and Treatment | South Africa | July 10, 2027 | |||
| 305483 | Anti-Cocaine Compositions and Treatment | Mexico | July 10, 2027 | |||
| 196411 | Mutants of Cocaine Esterase (CocE) Polypeptide, Nucleic Acids Encoding Them, Pharmaceutical Compositions Comprising Them and Uses Thereof | Israel | July 10, 2027 |
Sublingual CBP/Amitriptyline
| Patent No. | Title | Country / Region | Expiration Date | |||
| 6259452 | Compositions and Methods for Transmucosal Absorption | Japan | June 14, 2033 | |||
| 631144 | Compositions and Methods for Transmucosal Absorption | New Zealand | June 14, 2033 | |||
| I590820 | Compositions and Methods for Transmucosal Absorption | Taiwan R.O.C. | June 14, 2033 | |||
| 2013274003 | Compositions and Methods for Transmucosal Absorption | Australia | June 14, 2033 | |||
| I642429 | Compositions and Methods for Transmucosal Absorption | Taiwan R.O.C. | June 14, 2033 | |||
| 726488 | Compositions and Methods for Transmucosal Absorption | New Zealand | June 14, 2033 | |||
| I683660 | Compositions and Methods for Transmucosal Absorption | Taiwan R.O.C. | June 14, 2033 | |||
| 2018241128 | Compositions and Methods for Transmucosal Absorption | Australia | June 14, 2033 | |||
| 2876902 | Compositions and Methods for Transmucosal Absorption | Canada | June 14, 2033 | |||
| IDP000076019 | Compositions and Methods for Transmucosal Absorption | Indonesia | June 14, 2033 | |||
| 382516 | Compositions and Methods for Transmucosal Absorption | Mexico | June 14, 2033 | |||
2861223 (AL/P/2022/458 in Albania; P20221325T in Croatia; 602013082236.0 in Germany; 3111421 in Greece; 502022000069474 in Italy; P912561 in North Macedonia; SM-T-202200436 in San Marino; RS63822B1 in Serbia; and 2022-GE-787024 in Turkey)
|
Compositions and Methods for Transmucosal Absorption | European Patent Office – Italy, Albania, Austria, Belgium, Bulgaria, Cyprus, Czechia, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Iceland, Ireland, Latvia, Lithuania, Luxembourg, Malta, Monaco, Netherlands, Norway, Poland, Portugal, Romania, Slovakia, Slovenia, Spain, Sweden, Switzerland, United Kingdom, San Marino, Serbia, Croatia, North Macedonia and Turkey | June 14, 2033 | |||
| 236268 | Compositions for Transmucosal Delivery and Uses Thereof | Israel | June 14, 2033 | |||
| 2015/00288 | Compositions and Methods for Transmucosal Absorption | South Africa | June 14, 2033 | |||
| BR112014031394-6 | Compositions and Methods for Transmucosal Absorption | Brazil | June 14, 2033 | |||
| 1209361 | Compositions and Methods for Transmucosal Absorption | Hong Kong | June 14, 2033 | |||
| 398632 | Compositions and Methods for Transmucosal Absorption | Mexico | June 14, 2033 | |||
| A059897 | Compositions and Methods for Transmucosal Absorption | Venezuela | June 14, 2033 | |||
| MY-194495-A | Compositions and Methods for Transmucosal Absorption | Malaysia | June 14, 2033 | |||
| 3,118,913 | Compositions and Methods for Transmucosal Absorption | Canada | June 14, 2033 | |||
| 10201605407T | Compositions and Methods for Transmucosal Absorption | Singapore | June 14, 2033 |
30
CBP – Depression
| Patent No. | Title | Country / Region | Expiration Date | |||
| 11,998,516 | Methods and Compositions for Treating Depression Using Cyclobenzaprine | U.S.A. | March 5, 2032 | |||
| 2012225548 | Methods and Compositions for Treating Depression Using Cyclobenzaprine | Australia | March 6, 2032 | |||
| 2016222412 | Methods and Compositions for Treating Depression Using Cyclobenzaprine | Australia | March 6, 2032 | |||
| 2018204633 | Methods and Compositions for Treating Depression Using Cyclobenzaprine | Australia | March 6, 2032 | |||
| 2020203874 | Methods and Compositions for Treating Depression Using Cyclobenzaprine | Australia | March 6, 2032 | |||
| 614725 | Methods and Compositions for Treating Depression Using Cyclobenzaprine | New Zealand | March 6, 2032 | |||
| 714294 | Methods and Compositions for Treating Depression Using Cyclobenzaprine | New Zealand | March 6, 2032 | |||
| 2,829,200 | Methods and Compositions for Treating Depression Using Cyclobenzaprine | Canada | March 6, 2032 | |||
2683245
(AL/P/2020/15 in Albania; P20200142 in Croatia; MK/P/2020/68 in North Macedonia; 602012066717.6 in Germany; 3103147 in Greece; HU/E048596 in Hungary; 502020000014740 in Italy; 10476 in North Macedonia; SM-T-202000083 in San Marino; 60240 in Serbia; 2773834 in Spain; and 2020-GE-5216 in Turkey) |
Methods and Compositions for Treating Depression Using Cyclobenzaprine
|
European Patent Office – Albania, Austria, Belgium, Bulgaria, Switzerland, Cyprus, Czechia, Germany, Denmark, Estonia, Spain, Finland, France, United Kingdom, Greece, Croatia, Hungary, Ireland, Iceland, Italy, Lithuania, Luxembourg, Latvia, Monaco, Republic of North Macedonia, Malta, Netherlands, Norway, Poland, Portugal, Romania, Serbia, Sweden, Slovenia, Slovakia, San Marino, and Turkey | March 6, 2032 |
CBP – PTSD
| Patent No. | Title | Country / Region | Expiration Date | |||
| 9,918,948 | Methods and Compositions for Treating Symptoms Associated with Post-Traumatic Stress Disorder Using Cyclobenzaprine | U.S.A. | November 18, 2030 |
CBP Fatigue
| Patent No. | Title | Country / Region | Expiration Date | |||
| 9,474,728 | Methods and Compositions for Treating Fatigue Associated with Disordered Sleep Using Very Low Dose Cyclobenzaprine | U.S.A. | June 9, 2031 | |||
| 10,722,478 | Methods and Compositions for Treating Fatigue Associated with Disordered Sleep Using Very Low Dose Cyclobenzaprine | U.S.A. | June 9, 2031 |
31
CBP – Agitation in Neurodegenerative Condition
| Patent No. | Title | Country / Region | Expiration Date | |||
| 11,826,321 | Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions | U.S.A. | December 11, 2038 | |||
| 275289 | Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions | Israel | December 11, 2038 | |||
| 411601 | Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions | Mexico | December 11, 2038 | |||
| 3,083,341 | Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions | Canada | December 11, 2038 | |||
| 2020/03243 | Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions | South Africa | December 11, 2038 | |||
| MY-207073-A | Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions | Malaysia | December 11, 2038 | |||
| 2018383098 | Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions | Australia | December 11, 2038 |
CBP/Amitriptyline Eutectic Formulations
| Patent No. | Title | Country / Region | Expiration Date | |||
| 631152 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | New Zealand | March 14, 2034 | |||
| 747040 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | New Zealand | March 14, 2034 | |||
| 9,636,408 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | U.S.A. | March 14, 2034 | |||
| 9,956,188 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | U.S.A. | March 14, 2034 | |||
| 10,117,936 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | U.S.A. | March 14, 2034 | |||
| 10,322,094 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | U.S.A. | March 14, 2034 | |||
| 10,357,465 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | U.S.A. | September 18, 2035 | |||
| 10,736,859 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | U.S.A. | March 14, 2034 | |||
| 10,864,175 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | U.S.A. | March 14, 2034 | |||
| 10,864,176 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | U.S.A. | March 14, 2034 | |||
| 11,026,898 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | U.S.A. | September 18, 2035 | |||
| 11,737,991 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | U.S.A. | March 14, 2034 | |||
| 11,839,594 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | U.S.A. | March 14, 2034 | |||
| 6310542 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | Japan | March 14, 2034 | |||
| 6614724 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | Japan | September 18, 2035 | |||
| 6717902 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | Japan | September 18, 2035 |
32
| 6088 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | Saudi Arabia | March 14, 2034 | |||
| ZL201480024011.1 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | China | March 14, 2034 | |||
| ZL.201580050140.2 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | China | September 18, 2035 | |||
| 2014233277 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | Australia | March 14, 2034 | |||
| 2015317336 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | Australia | September 18, 2035 | |||
| I661825 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | Taiwan R.O.C. | March 14, 2034 | |||
| I740136 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | Taiwan R.O.C. | March 14, 2034 | |||
| IDP000055516 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | Indonesia | March 14, 2034 | |||
| IDP000063221 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | Indonesia | September 18, 2035 | |||
| IDP000076872 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | Indonesia | March 14, 2034 | |||
2968992 (1211591 in Austria, CZ2014-762323 in Czechia, 602014058260.5 in Germany, E018723 in Estonia, P20200055 in Croatia, 201361792757 P in Ireland, 2020.67 in Monaco, P-2020/0094 in Serbia, 201431487 in Slovenia, 33269 in Slovakia, 2020000045 in San Marino, AL/P/2019/906 in Albania, MK/P/2020/67 in Republic of North Macedonia, 3102655 in Greece, 502020000007756 in Italy)
|
Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | European Patent Office - Albania, Austria, Belgium, Bulgaria, Croatia, Cyprus, Czechia, Denmark, Estonia, Finland, France, Republic of North Macedonia, Germany, Greece, Hungary, Iceland, Ireland, Italy, Latvia, Lithuania, Luxembourg, Malta, Monaco, Netherlands, Norway, Poland, Portugal, Romania, San Marino, Serbia, Slovakia, Slovenia, Spain, Sweden, Switzerland, Turkey, United Kingdom | March 14, 2034
| |||
| 3650081 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | European Patent Office - Albania, Austria, Belgium, Bulgaria, Croatia, Cyprus, Czechia, Denmark, Estonia, Finland, France, Republic of North Macedonia, Germany, Greece, Hungary, Iceland, Ireland, Italy, Latvia, Lithuania, Luxembourg, Malta, Monaco, Netherlands, Norway, Poland, Portugal, Romania, San Marino, Serbia, Slovakia, Slovenia, Spain, Sweden, Switzerland, Turkey, United Kingdom | March 14, 2034 |
33
| 241353 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | Israel | March 14, 2034 | |||
| 251218 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Methods of Producing Same | Israel | September 18, 2035 | |||
| 277814 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Methods of Producing Same | Israel | September 18, 2034 | |||
| 370021 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | Mexico | March 14, 2034 | |||
| 387402 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | Mexico | September 18, 2035 | |||
| 388137 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | Mexico | March 14, 2034 | |||
| 2015/07443 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | South Africa | March 14, 2034 | |||
| 2017/01637 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | South Africa | September 18, 2035 | |||
| BR112015022095-9 | Pharmaceutical Composition, Method of Fabrication, Eutectic Composition and Use of Compositions Containing Cyclobenzaprine HCl and Mannitol | Brazil | March 14, 2034 | |||
| 2904812 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | Canada | March 14, 2034 | |||
| 3119755 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | Canada | March 14, 2034 | |||
| 2,961,822 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | Canada | September 18, 2035 | |||
| HK1218727 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | Hong Kong | March 14, 2034 | |||
| MY-186047-A | Eutectic Formulations of Cyclobenzaprine Hydrochloride | Malaysia | September 18, 2035 | |||
| 398845 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | India | September 18, 2035 | |||
| 441374 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | India | March 14, 2034 | |||
| MY-196014-A | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | Malaysia | March 14, 2034 | |||
| ZL201910263541.6 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | China | March 14, 2034 | |||
| 2020289838 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | Australia | September 18, 2035 | |||
| HK40047283 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | Hong Kong | September 18, 2035 | |||
| ZL202011576351.9 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | China | September 18, 2035 | |||
| 730379 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | New Zealand | September 18, 2035 | |||
| 768064 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | New Zealand | September 18, 2035 | |||
| 40013124 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | Hong Kong | March 14, 2034 | |||
| 40030559 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | Hong Kong | March 14, 2034 |
Analogs of CBP
| Patent No. | Title | Country / Region | Expiration Date | |||
| 11,517,557 | Analogs of Cyclobenzaprine and Amitryptilene | U.S.A. | July 13, 2038 | |||
| 12,156,864 | Analogs of Cyclobenzaprine and Amitryptilene | U.S.A. | July 13, 2038 | |||
| 7330964 | Analogs of Cyclobenzaprine and Amitryptilene | Japan | July 13, 2038 |
34
Oxytocin therapeutics
| Patent No. | Title | Country / Region | Expiration Date | |||
| 9,629,894 | Magnesium-Containing Oxytocin Formulations and Methods of Use | U.S.A. | January 7, 2036 | |||
| 11,389,473 | Magnesium-Containing Oxytocin Formulations and Methods of Use | U.S.A. | January 7, 2036 | |||
| 11201705591P | Magnesium-Containing Oxytocin Formulations and Methods of Use | Singapore | January 7, 2036 | |||
| 388286 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Mexico | January 7, 2036 | |||
| 253347 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Israel | January 7, 2036 | |||
| 7030517 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Japan | January 7, 2036 | |||
| ZL201680013809.5 | Magnesium-Containing Oxytocin Formulations and Methods of Use | China | January 7, 2036 | |||
3242676 (P20231438 in Croatia; 602016083177.5 in Germany; 3114323 in Greece; HU/E065385 in Hungary; 65034 in Serbia; SM-T-202400020 in San Marino; and 2023-GE-778438 in Turkey)
|
Magnesium-Containing Oxytocin Formulations and Methods of Use | Europe – (Albania, Austria, Belgium, Bulgaria, Croatia, Cyprus, Czechia, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Iceland, Ireland, Italy, Latvia, Lithuania, Luxembourg, Malta, Monaco, Netherlands, North Macedonia, Norway, Poland, Portugal, Romania, San Marino, Serbia, Slovakia, Slovenia, Spain, Sweden, Switzerland, Turkey, United Kingdom) | January 7, 2036 | |||
| 2017/05176 | Magnesium-Containing Oxytocin Formulations and Methods of Use | South Africa | January 7, 2036 | |||
| 1252942 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Hong Kong | January 7, 2036 | |||
| 2020286221 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Australia | January 7, 2036 | |||
| 734097 | Magnesium-Containing Oxytocin Formulations and Methods of Use | New Zealand | January 7, 2036 | |||
| 771693 | Magnesium-Containing Oxytocin Formulations and Methods of Use | New Zealand | January 7, 2036 | |||
| 10-2677904 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Republic of Korea | January 7, 2036 | |||
| 7455402 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Japan | January 7, 2036 | |||
| BR112017014545-6 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Brazil | January 7, 2036 | |||
| BR122024008322-1 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Brazil | January 7, 2036 | |||
| 7756953 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Japan | January 7, 2036 |
35
| 11,970,554 | Labeled Oxytocin and Method of Manufacture and Use | U.S.A. | March 1, 2039 | |||
| 12,466,855 | Labeled Oxytocin and Method of Manufacture and Use | U.S.A. | March 1, 2039 | |||
| 7093559 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Japan | April 12, 2037 | |||
| 2017250505 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Australia | April 12, 2037 | |||
| 2023203831 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Australia | April 12, 2037 | |||
| ZL201780036185.3 | Magnesium-Containing Oxytocin Formulations and Methods of Use | China | April 12, 2037 | |||
| 40005263 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Hong Kong | April 12, 2037 | |||
| 747221 | Magnesium-Containing Oxytocin Formulations and Methods of Use | New Zealand | April 12, 2037 | |||
| 787097 | Magnesium-Containing Oxytocin Formulations and Methods of Use | New Zealand | April 12, 2037 | |||
| 3,020,179 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Canada | April 12, 2037 | |||
| 12,156,897 | Magnesium-Containing Oxytocin Formulations and Methods of Use | U.S.A. | April 12, 2037 | |||
| 417307 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Mexico | April 12, 2037 | |||
3442560 (602017086259.2 in Germany; and SM/T/2025/000041 in San Marino)
|
Magnesium-Containing Oxytocin Formulations and Methods of Use | Europe – (Albania, Austria, Belgium, Bulgaria, Croatia, Cyprus, Czechia, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Iceland, Ireland, Italy, Latvia, Lithuania, Luxembourg, Malta, Monaco, Netherlands, North Macedonia, Norway, Poland, Portugal, Romania, San Marino, Serbia, Slovakia, Slovenia, Spain, Sweden, Switzerland, Turkey, and United Kingdom) | April 12, 2037 | |||
2575853 (2595251 in Spain) |
Methods and Pharmaceutical Composition for the Treatment of a Feeding Disorder with Early-Onset in a Patient | Europe – (Spain, France, Germany, Italy, and United Kingdom) | May 25, 2031 | |||
| 8,853,158 | Methods for the Treatment of a Feeding Disorder with Onset During Neonate Development Using an Agonist of the Oxytocin Receptor | U.S.A. | May 25, 2031 | |||
| 9,125,862 | Methods for the Treatment of Prader-Willi-like Syndrome or Non-Organic Failure to Thrive (NOFITT) Feeding Disorder Using an Agonist of the Oxytocin Receptor | U.S.A. | May 25, 2031 | |||
2571511 (2526672 in Spain) |
New Uses of Oxytocin-like Molecules and Related Methods | Europe – (Switzerland, Spain, France, Germany, Italy, United Kingdom, and Ireland) | May 17, 2031 | |||
| 9,101,569 | Methods for the Treatment of Insulin Resistance | U.S.A. | June 22, 2031 |
36
Nociceptin/Orphanin FQ therapeutics
| Patent No. | Title | Country / Region | Expiration Date | |||
| 8,551,949 | Methods for treatment of pain | U.S.A. | August 11, 2031 | |||
| 9,238,053 | Methods for treatment of pain | U.S.A. | October 12, 2030 | |||
| 2010281436 | Methods for treatment of pain | Australia | July 27, 2030 | |||
| ZL 201080042858.4 | Methods for treatment of pain | China | July 27, 2030 | |||
| 2459183 (602010028120.5 in Germany) | Methods for treatment of pain | Europe – (Switzerland, Germany, Denmark, France, and United Kingdom) | July 27, 2030 | |||
| 1169804 | Methods for treatment of pain | Hong Kong | July 27, 2030 | |||
| 329837 | Methods for treatment of pain | Mexico | July 27, 2030 | |||
| 597763 | Methods for treatment of pain | New Zealand | July 27, 2030 | |||
| 10201406930U | Methods for treatment of pain | Singapore | July 27, 2030 | |||
| 201200584 | Methods for treatment of pain | South Africa | July 27, 2030 | |||
| 2,769,347 | Methods for treatment of pain | Canada | July 27, 2030 | |||
| 413642 | Methods for treatment of pain | India | July 27, 2030 |
Tianeptine – Neurocognitive Dysfunction
| Patent No. | Title | Country / Region | Expiration Date | |||
| 9,314,469 | Method for Treating Neurocognitive Dysfunction | U.S.A. | September 24, 2030 | |||
| 2723688 | Method for Treating Neurodegenerative Dysfunction | Canada | April 30, 2029 | |||
| 2299822 (602009047361.1 in Germany; 2644511 in Spain; and E911827 in Austria) | Method for Treating Neurodegenerative Dysfunction | European Patent Office – Austria, Belgium, Switzerland, Germany, Spain, France, United Kingdom, Ireland, Luxembourg, Monaco, and Portugal | April 30, 2029 | |||
| 3246031 (602009057284.9 in Germany; 2727851 in Spain; and E1100344 in Austria) | Method for Treating Neurocognitive Dysfunction | European Patent Office – Austria, Belgium, Switzerland, Germany, Spain, France, United Kingdom, Ireland, Luxembourg, Monaco, and Portugal | April 30, 2029 |
TFF2 therapeutics
| Patent No. | Title | Country / Region | Expiration Date | |||
| 10,124,037 | Trefoil family factor proteins and uses thereof | U.S.A | April 2, 2033 | |||
| 11,167,010 | Trefoil family factor proteins and uses thereof | U.S.A | April 2, 2033 |
37
Synthetic Chimeric Poxviruses
| Patent No. | Title | Country / Region | Expiration Date | |||
| 11,345,896 | Synthetic Chimeric Poxviruses | U.S.A | November 2, 2037 | |||
| 12,365,879 | Synthetic Chimeric Poxviruses | U.S.A | November 2, 2037 | |||
| 397516 | Synthetic Chimeric Poxviruses | Mexico | November 2, 2037 | |||
| 2019/02868 | Synthetic Chimeric Poxviruses | South Africa | November 2, 2037 | |||
| MY-200354-A | Synthetic Chimeric Poxviruses | Malaysia | November 2, 2037 | |||
| 2017353868 | Synthetic Chimeric Poxviruses | Australia | November 2, 2037 | |||
| ZL201780078546.0 | Synthetic Chimeric Poxviruses | China | November 2, 2037 | |||
| 40014109 | Synthetic Chimeric Poxviruses | Hong Kong | November 2, 2037 | |||
| 2022/04981 | Synthetic Chimeric Poxviruses | South Africa | November 2, 2037 | |||
| 2024/03393 | Synthetic Chimeric Poxviruses | South Africa | November 2, 2037 | |||
| IDP000098405 | Synthetic Chimeric Poxviruses | Indonesia | November 2, 2037 | |||
| 266399 | Synthetic Chimeric Poxviruses | Israel | November 2, 2037 | |||
| I887191 | Synthetic Chimeric Poxviruses | Taiwan | November 2, 2037 | |||
| 752893 | Synthetic Chimeric Poxviruses | New Zealand | November 2, 2037 |
Synthetic Vaccinia Virus
| Patent No. | Title | Country / Region | Expiration Date | |||
| 12,529,036 | Synthetic Chimeric Vaccinia Virus | U.S.A | May 2, 2039 | |||
| ZL201980029677.9 | Synthetic Chimeric Vaccinia Virus | China | May 2, 2039 | |||
| 431454 | Synthetic Chimeric Vaccinia Virus | Mexico | May 2, 2039 | |||
| 2019262149 | Synthetic Chimeric Vaccinia Virus | Australia | May 2, 2039 | |||
| HK40047064 | Synthetic Chimeric Vaccinia Virus | Hong Kong | May 2, 2039 | |||
| MY-212174-A | Synthetic Chimeric Vaccinia Virus | Malaysia | May 2, 2039 |
Poxvirus vaccine against COVID-19
| Patent No. | Title | Country / Region | Expiration Date | |||
| I902763 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | Taiwan | February 16, 2041 |
Triptan Compound – Formulations
| Patent No. | Title | Country / Region | Expiration Date | |||
| 9,211,282 | Formulations Comprising Triptan Compounds | U.S.A | July 19, 2031 | |||
| 9,610,280 | Formulations Comprising Triptan Compounds | U.S.A | June 16, 2030 | |||
| 9,974,770 | Formulations Comprising Triptan Compounds | U.S.A | June 16, 2030 | |||
| 10,603,305 | Formulations Comprising Triptan Compounds | U.S.A | June 16, 2030 | |||
| 11,337,962 | Formulations Comprising Triptan Compounds | U.S.A. | June 16, 2030 | |||
| 12,090,139 | Formulations Comprising Triptan Compounds | U.S.A. | June 16, 2030 | |||
| 2010299607 | Formulations Comprising Triptan Compounds | Australia | June 17, 2030 | |||
| 2775404 | Formulations Comprising Triptan Compounds | Canada | June 17, 2030 | |||
2480197
(E760080 in Austria; 502016000000073 in Italy; 602010028995.8 in Germany; and 2553862 in Spain)
|
Formulations Comprising Triptan Compounds | European Patent Office - Austria, Belgium, Czechia, Denmark, France, Germany, Italy, Spain, Switzerland, and United Kingdom | June 17, 2030 | |||
| 5845183 | Formulations Comprising Triptan Compounds | Japan | June 17, 2030 | |||
| 101646079 | Formulations Comprising Triptan Compounds | Republic of Korea | June 17, 2030 | |||
| 338110 | Formulations Comprising Triptan Compounds | Mexico | June 17, 2030 | |||
| 599344 | Formulations Comprising Triptan Compounds | New Zealand | June 17, 2030 | |||
| 2012/02168 | Formulations Comprising Triptan Compounds | South Africa | June 17, 2030 |
38
Triptan Compound – Migraine
| Patent No. | Title | Country / Region | Expiration Date | |||
| 10,537,554 | Pharmaceutical Composition for Treating Migraine | U.S.A | January 29, 2036 | |||
| 11,364,224 | Pharmaceutical Composition for Treating Migraine | U.S.A | January 29, 2036 | |||
| 12,097,183 | Pharmaceutical Composition for Treating Migraine | U.S.A | January 29, 2036 | |||
| 385725 | Pharmaceutical Composition Comprising Sumatriptan for Treating Migraine | Mexico | February 1, 2036 | |||
| 2994748 | Pharmaceutical Composition Comprising Sumatriptan for Treating Migraine | Canada | February 1, 2036 | |||
CD40 and anti-CD154 Therapeutics
| Patent No. | Title | Country / Region | Expiration Date | |||
3993876
(E1717312 in Austria; P20241602 in Croatia; 602020036714.4 in Germany; 3116977 in Greece; HU/E069680 in Hungary; 502024000056334 in Italy; 66218 in Serbia; 2994684 in Spain; and SM-T-202400527 in San Marino)
|
Anti-CD154 antibodies and uses thereof | Europe – (Albania, Austria, Belgium, Bulgaria, Croatia, Cyprus, Czechia, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Iceland, Ireland, Italy, Latvia, Lithuania, Luxembourg, Malta, Monaco, Netherlands, North Macedonia, Norway, Poland, Portugal, Romania, San Marino, Serbia, Slovenia, Slovakia, Spain, Sweden, Switzerland, Turkey, United Kingdom) | July 1, 2040 | |||
| ZL202080059891.1 | Anti-CD154 antibodies and uses thereof | China | July 1, 2040 | |||
| 40074747 | Anti-CD154 antibodies and uses thereof | Hong Kong | July 1, 2040 | |||
| 7775080 | Anti-CD154 antibodies and uses thereof | Japan | July 1, 2040 | |||
| MY-209630-A | Anti-CD154 antibodies and uses thereof | Malaysia | July 1, 2040 | |||
| 2022/01378 | Anti-CD154 antibodies and uses thereof | South Africa | July 1, 2040 |
CBP - ASD and PTSD
| Patent No. | Title | Country / Region | Expiration Date | |||
| 420368 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | Mexico | August 20, 2039 | |||
| 2019323764 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | Australia | August 20, 2039 | |||
| 7691739 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | Japan | August 20, 2039 |
CBP – Sexual dysfunction
| Patent No. | Title | Country / Region | Expiration Date | |||
| ZL202180040673.8 | Cyclobenzaprine Treatment for Sexual Dysfunction | China | ||||
| 40090080 | Cyclobenzaprine Treatment for Sexual Dysfunction | Hong Kong | ||||
| 7780447 | Cyclobenzaprine Treatment for Sexual Dysfunction | Japan |
39
Pending Patent Applications
Our current pending patent applications are as follows:
CD40 and anti-CD154 Therapeutics
| Application No. | Title | Country / Region | ||
| 17/623,710 | Anti-CD154 antibodies and uses thereof | U.S.A. | ||
| 2020300002 | Anti-CD154 antibodies and uses thereof | Australia | ||
| BR112021026410-8 | Anti-CD154 antibodies and uses thereof | Brazil | ||
| BR122024022801-7 | Anti-CD154 antibodies and uses thereof | Brazil | ||
| 3145453 | Anti-CD154 antibodies and uses thereof | Canada | ||
| 202510443183.2 | Anti-CD154 antibodies and uses thereof | China | ||
| 24195755.4 | Anti-CD154 antibodies and uses thereof | European Patent Office | ||
| 202217004870 | Anti-CD154 antibodies and uses thereof | India | ||
| P00202200763 | Anti-CD154 antibodies and uses thereof | Indonesia | ||
| 289354 | Anti-CD154 antibodies and uses thereof | Israel | ||
| 2025-97959 | Anti-CD154 antibodies and uses thereof | Japan | ||
| PI 2024006778 | Anti-CD154 antibodies and uses thereof | Malaysia | ||
| MX/a/2022/000133 | Anti-CD154 antibodies and uses thereof | Mexico | ||
| 784548 | Anti-CD154 antibodies and uses thereof | New Zealand | ||
| 822935 | Anti-CD154 antibodies and uses thereof | New Zealand | ||
| 11202114433Y | Anti-CD154 antibodies and uses thereof | Singapore | ||
| 2024/09160 | Anti-CD154 antibodies and uses thereof | South Africa | ||
| 42025108505.6 | Anti-CD154 antibodies and uses thereof | Hong Kong | ||
| 18/271,098 | Methods of Inducing Immune Tolerance with Modified Anti-CD154 Antibodies | U.S.A. | ||
| 2022205313 | Methods of Inducing Immune Tolerance with Modified Anti-CD154 Antibodies | Australia | ||
| BR112023013285-1 | Methods of Inducing Immune Tolerance with Modified Anti-CD154 Antibodies | Brazil | ||
| 3207098 | Methods of Inducing Immune Tolerance with Modified Anti-CD154 Antibodies | Canada | ||
| 202280019221.6 | Methods of Inducing Immune Tolerance with Modified Anti-CD154 Antibodies | China | ||
| 22701768.8 | Methods of Inducing Immune Tolerance with Modified Anti-CD154 Antibodies | European Patent Office | ||
| P00202307159 | Methods of Inducing Immune Tolerance with Modified Anti-CD154 Antibodies | Indonesia | ||
| 304253 | Methods of Inducing Immune Tolerance with Modified Anti-CD154 Antibodies | Israel | ||
| 2023-541043 | Methods of Inducing Immune Tolerance with Modified Anti-CD154 Antibodies | Japan | ||
| PI 2023003993 | Methods of Inducing Immune Tolerance with Modified Anti-CD154 Antibodies | Malaysia | ||
| MX/a/2023/008055 | Methods of Inducing Immune Tolerance with Modified Anti-CD154 Antibodies | Mexico | ||
| 801414 | Methods of Inducing Immune Tolerance with Modified Anti-CD154 Antibodies | New Zealand | ||
| 11202305000R | Methods of Inducing Immune Tolerance with Modified Anti-CD154 Antibodies | Singapore | ||
| 2023/06791 | Methods of Inducing Immune Tolerance with Modified Anti-CD154 Antibodies | South Africa | ||
| 62024090562.5 | Methods of Inducing Immune Tolerance with Modified Anti-CD154 Antibodies | Hong Kong | ||
| 62024091450.2 | Methods of Inducing Immune Tolerance with Modified Anti-CD154 Antibodies | Hong Kong | ||
| PCT/EP2025/065085 | Treatment Methods Comprising Administration of Modified CD154 Antibodies | PCT |
40
CBP/Amitriptyline Eutectic Formulations
| Application No. | Title | Country / Region | ||
| 19/230,858 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | U.S.A. | ||
| BR112017005231-8 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | Brazil | ||
| BR122020020968-2 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | Brazil | ||
| 15841528.1 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | European Patent Office | ||
| 18101200.4 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | Hong Kong | ||
| 2023-188486 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | Japan | ||
| 2025-76812 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | Japan |
| Application No. | Title | Country / Region | ||
| PI 2023000078 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | Malaysia | ||
| 517381123 | Eutectic Formulations of Cyclobenzaprine Hydrochloride | Saudi Arabia | ||
| 10201707528W | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride (Allowed) | Singapore | ||
| 10201902203V | Eutectic Formulations of Cyclobenzaprine Hydrochloride | Singapore | ||
| 2014-000391 | Eutectic Formulations of Cyclobenzaprine Hydrochloride and Amitriptyline Hydrochloride | Venezuela |
Sublingual CBP/Amitriptyline
| Application No. | Title | Country / Region | ||
| 19/409,704 | Compositions and Methods for Transmucosal Absorption | U.S.A. | ||
| 20230100254 | Compositions and Methods for Transmucosal Absorption | Argentina | ||
| 202010024102.2 | Compositions and Methods for Transmucosal Absorption | China | ||
| 2013/24661 | Compositions and Methods for Transmucosal Absorption | Gulf Cooperation Council | ||
| 2013/37088 | Compositions and Methods for Transmucosal Absorption | Gulf Cooperation Council | ||
| 2013/40660 | Compositions and Methods for Transmucosal Absorption | Gulf Cooperation Council | ||
| 42020020336.2 | Compositions and Methods for Transmucosal Absorption | Hong Kong | ||
| P-00 2021 01421 | Compositions and Methods for Transmucosal Absorption | Indonesia | ||
| 2024-14696 | Compositions and Methods for Transmucosal Absorption | Japan | ||
| 10202401383X | Compositions and Methods for Transmucosal Absorption | Singapore |
CBP – Agitation in Neurodegenerative Condition
| Application No. | Title | Country / Region | ||
| BR112020011345-0 | Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions | Brazil | ||
| 201880079917.1 | Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions | China | ||
| 18847270.8 | Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions | European Patent Office | ||
| P00202004178 | Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions | Indonesia | ||
| 202017023747 | Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions | India | ||
| 62020022462.9 | Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions | Hong Kong | ||
| 62021029558.5 | Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions | Hong Kong | ||
| 42024101171.7 | Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions | Hong Kong | ||
| 10202303446R | Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions | Singapore | ||
| 2026-6717 | Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions | Japan | ||
| 202410396685.X | Cyclobenzaprine Treatment for Agitation, Psychosis and Cognitive Decline in Dementia and Neurodegenerative Conditions | China |
41
Analogs of CBP
| Application No. | Title | Country / Region | ||
18/950,382 |
Analogs of Cyclobenzaprine and Amitriptyline |
U.S.A. | ||
| 3069699 | Analogs of Cyclobenzaprine and Amitriptyline | Canada | ||
| 201880050758.2 | Analogs of Cyclobenzaprine and Amitriptyline | China | ||
| 18831505.5 | Analogs of Cyclobenzaprine and Amitriptyline | European Patent Office | ||
| 2024-027046 | Analogs of Cyclobenzaprine and Amitriptyline | Japan |
CBP – ASD and PTSD
| Application No. | Title | Country / Region | ||
| 2019/38140 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | Gulf Cooperation Council | ||
| 108129709 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | Taiwan R.O.C. | ||
| 17/269,106 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | U.S.A. | ||
| PI2021000802 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | Malaysia | ||
| 772889 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | New Zealand | ||
| 813830 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | New Zealand | ||
| BR112021003107-3 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | Brazil | ||
| 3109258 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | Canada | ||
| 201980062283.3 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | China | ||
| 202510112547.9 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | China | ||
| 19802247.7 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | European Patent Office | ||
| 62021045278.0 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | Hong Kong | ||
| 62022046260.5 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | Hong Kong | ||
| 42025114696.5 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | Hong Kong | ||
| 202117011223 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | India | ||
| P00202101716 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | Indonesia | ||
| 280921 | Cyclobenzaprine or Amitriptyline Containing Compositions for Use in Treating Stress Disorders | Israel | ||
| 2025-32822 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | Japan | ||
| 10-2024-02502Q | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | Singapore | ||
| 2021/01121 | Methods of Treating Acute Stress Disorder and Posttraumatic Stress Disorder | South Africa |
CBP – Fibromyalgia
| Application No. | Title | Country / Region | ||
| 18/265,525 | Cyclobenzaprine Treatment for Fibromyalgia | U.S.A. | ||
| 19/421,039 | Cyclobenzaprine Treatment for Fibromyalgia | U.S.A. | ||
| 2021396509 | Cyclobenzaprine Treatment for Fibromyalgia | Australia | ||
| 3204202 | Cyclobenzaprine Treatment for Fibromyalgia | Canada | ||
| 202180089897.8 | Cyclobenzaprine Treatment for Fibromyalgia | China | ||
| 21844438.8 | Cyclobenzaprine Treatment for Fibromyalgia | European Patent Office | ||
| 62024089847.3 | Cyclobenzaprine Treatment for Fibromyalgia | Hong Kong | ||
| 62024090216.8 | Cyclobenzaprine Treatment for Fibromyalgia | Hong Kong | ||
| 202317044026 | Cyclobenzaprine Treatment for Fibromyalgia | India | ||
| P00202306147 | Cyclobenzaprine Treatment for Fibromyalgia | Indonesia | ||
| 303497 | Cyclobenzaprine Treatment for Fibromyalgia | Israel | ||
| 2023-542924 | Cyclobenzaprine Treatment for Fibromyalgia | Japan | ||
| PI 2023003286 | Cyclobenzaprine Treatment for Fibromyalgia | Malaysia | ||
| MX/a/2023/006720 | Cyclobenzaprine Treatment for Fibromyalgia | Mexico | ||
| 800700 | Cyclobenzaprine Treatment for Fibromyalgia | New Zealand | ||
| 523441103 | Cyclobenzaprine Treatment for Fibromyalgia | Saudi Arabia | ||
| 10202403823V | Cyclobenzaprine Treatment for Fibromyalgia | Singapore | ||
| 2023/06139 | Cyclobenzaprine Treatment for Fibromyalgia | South Africa |
42
CBP – Fibromyalgia (Early Onset Response, Favorable Tolerability, and Side Effect Profile)
| Application No. | Title | Country / Region | ||
| PCT/US2024/061125 | Early Onset Response, Favorable Tolerability, and Side Effect Profile in the Treatment of Fibromyalgia | PCT | ||
| 18/988,194 | Early Onset Response, Favorable Tolerability, and Side Effect Profile in the Treatment of Fibromyalgia | U.S.A. |
CBP – Acute Stress Reaction or Acute Stress Disorder
| Application No. | Title | Country / Region | ||
| PCT/US2025/012803 | Cyclobenzaprine Treatment for Acute Stress Reaction or Acute Stress Disorder | PCT |
CBP – Alcohol Use Disorder
| Application No. | Title | Country / Region | ||
| 18/037,815 | Cyclobenzaprine Treatment for Alcohol Use Disorder | U.S.A. | ||
| 2021382668 | Cyclobenzaprine Treatment for Alcohol Use Disorder | Australia | ||
| 112023009731-2 | Cyclobenzaprine Treatment for Alcohol Use Disorder | Brazil | ||
| 3202722 | Cyclobenzaprine Treatment for Alcohol Use Disorder | Canada | ||
| 202180088339.X | Cyclobenzaprine Treatment for Alcohol Use Disorder | China | ||
| 21827298.7 | Cyclobenzaprine Treatment for Alcohol Use Disorder | European Patent Office | ||
| 62024087606.5 | Cyclobenzaprine Treatment for Alcohol Use Disorder | Hong Kong | ||
| 62024089500.8 | Cyclobenzaprine Treatment for Alcohol Use Disorder | Hong Kong | ||
| 202317038485 | Cyclobenzaprine Treatment for Alcohol Use Disorder | India | ||
| 303050 | Cyclobenzaprine Treatment for Alcohol Use Disorder | Israel | ||
| 2023-530204 | Cyclobenzaprine Treatment for Alcohol Use Disorder | Japan | ||
| MX/a/2023/005899 | Cyclobenzaprine Treatment for Alcohol Use Disorder | Mexico | ||
| 800112 | Cyclobenzaprine Treatment for Alcohol Use Disorder | New Zealand | ||
| 10202501271X | Cyclobenzaprine Treatment for Alcohol Use Disorder | Singapore | ||
| 2023/05747 | Cyclobenzaprine Treatment for Alcohol Use Disorder | South Africa |
CBP – Sexual dysfunction
| Application No. | Title | Country / Region | ||
| 17/226,058 | Cyclobenzaprine Treatment for Sexual Dysfunction | U.S.A | ||
| 2021253592 | Cyclobenzaprine Treatment for Sexual Dysfunction | Australia | ||
| 3179754 | Cyclobenzaprine Treatment for Sexual Dysfunction | Canada | ||
| 21721779.3 | Cyclobenzaprine Treatment for Sexual Dysfunction (Allowed) | European Patent Office | ||
| 2025-172812 | Cyclobenzaprine Treatment for Sexual Dysfunction | Japan | ||
| 62023077251.4 | Cyclobenzaprine Treatment for Sexual Dysfunction | Hong Kong | ||
| 62025103395.2 | Cyclobenzaprine Treatment for Sexual Dysfunction | Hong Kong | ||
| 18/698,483 | Cyclobenzaprine for Treatment or Prevention of Sexual Dysfunction Associated with Mental Health Conditions in Female Patients | U.S.A. | ||
| 22800445.3 | Cyclobenzaprine for Treatment or Prevention of Sexual Dysfunction Associated with Mental Health Conditions in Female Patients | European Patent Office |
43
CBP – Post-Acute Sequelae of SARS-CoV-2 (PASC)
| Application No. | Title | Country / Region | ||
| 18/212,500 | Cyclobenzaprine Treatment for Post-Acute Sequelae of (SARS)-CoV-2 Infection (PASC) | U.S.A. | ||
| 2023286504 | Cyclobenzaprine Treatment for Post-Acute Sequelae of (SARS)-CoV-2 Infection (PASC) | Australia | ||
| 36260042 | Cyclobenzaprine Treatment for Post-Acute Sequelae of (SARS)-CoV-2 Infection (PASC) | Canada | ||
| 202380055899.4 | Cyclobenzaprine Treatment for Post-Acute Sequelae of (SARS)-CoV-2 Infection (PASC) | China | ||
| 23744274.4 | Cyclobenzaprine Treatment for Post-Acute Sequelae of (SARS)-CoV-2 Infection (PASC) | European Patent Office | ||
| 62025114250.6 | Cyclobenzaprine Treatment for Post-Acute Sequelae of (SARS)-CoV-2 Infection (PASC) | Hong Kong | ||
| 62025114926.1 | Cyclobenzaprine Treatment for Post-Acute Sequelae of (SARS)-CoV-2 Infection (PASC) | Hong Kong | ||
| 317836 | Cyclobenzaprine Treatment for Post-Acute Sequelae of (SARS)-CoV-2 Infection (PASC) | Israel | ||
| 2024-575083 | Cyclobenzaprine Treatment for Post-Acute Sequelae of (SARS)-CoV-2 Infection (PASC) | Japan | ||
| MX/a/2025/000142 | Cyclobenzaprine Treatment for Post-Acute Sequelae of (SARS)-CoV-2 Infection (PASC) | Mexico | ||
| 817589 | Cyclobenzaprine Treatment for Post-Acute Sequelae of (SARS)-CoV-2 Infection (PASC) | New Zealand | ||
| 2025/00293 | Cyclobenzaprine Treatment for Post-Acute Sequelae of (SARS)-CoV-2 Infection (PASC) | South Africa |
Oxytocin therapeutics
| Application No. | Title | Country / Region | ||
| 2024203428 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Australia | ||
| 2972975 | Magnesium-Containing Oxytocin Formulations and Methods of Use (Allowed) | Canada | ||
| 42024096348.8 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Hong Kong | ||
| 23201255.9 | Magnesium-Containing Oxytocin Formulations and Methods of Use | European Patent Office | ||
| 2025-165798 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Japan | ||
| 19710979.6 | Labeled Oxytocin and Method of Manufacture and Use | European Patent Office | ||
| 2025-230634 | Labeled Oxytocin and Method of Manufacture and Use | Japan | ||
| 2025-230635 | Labeled Oxytocin and Method of Manufacture and Use | Japan | ||
| 18/921,777 | Magnesium-Containing Oxytocin Formulations and Methods of Use | U.S.A. | ||
| 2023100344997 | Magnesium-Containing Oxytocin Formulations and Methods of Use | China | ||
| 24214056.4 | Magnesium-Containing Oxytocin Formulations and Methods of Use | European Patent Office | ||
| 2024-196703 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Japan | ||
| 2025256187 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Australia | ||
| 42023079422.4 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Hong Kong | ||
| 42025113914.3 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Hong Kong | ||
| MX/a/2023/008840 | Magnesium-Containing Oxytocin Formulations and Methods of Use | Mexico | ||
| PCT/US2025/020483 | Oxytocin Peptide Analogs | PCT |
44
Nociceptin/Orphanin FQ therapeutics
| Application No. | Title | Country / Region | ||
| BR122021007932-3 | Methods for Treatment of Pain | Brazil | ||
Synthetic Chimeric Poxviruses
| Application No. | Title | Country / Region | ||
| 19/249,853 | Synthetic Chimeric Poxviruses | U.S.A. | ||
| 19/249,868 | Synthetic Chimeric Poxviruses | U.S.A. | ||
| P 20170103043 | Synthetic Chimeric Poxviruses | Argentina | ||
| 2017/34209 | Synthetic Chimeric Poxviruses | Gulf Cooperation Council | ||
| 2017/41626 | Synthetic Chimeric Poxviruses | Gulf Cooperation Council | ||
| BR112019008781-8 | Synthetic Chimeric Poxviruses | Brazil | ||
| BR112019008781-9 | Synthetic Chimeric Poxviruses | Brazil | ||
| 3,042,694 | Synthetic Chimeric Poxviruses | Canada | ||
| 17868045.0 | Synthetic Chimeric Poxviruses | European Patent Office | ||
| 201917021814 | Synthetic Chimeric Poxviruses | India | ||
319942 |
Synthetic Chimeric Poxviruses |
Israel | ||
| 2019-545700 | Synthetic Chimeric Poxviruses | Japan | ||
| 2024-93677 | Synthetic Chimeric Poxviruses | Japan | ||
| 11201903893P | Synthetic Chimeric Poxviruses (Allowed) | Singapore | ||
| 2025/03540 | Synthetic Chimeric Poxviruses | South Africa | ||
| 2017-000418 | Synthetic Chimeric Poxviruses | Venezuela | ||
| 62020003684.1 | Synthetic Chimeric Poxviruses | Hong Kong | ||
| 792675 | Synthetic Chimeric Poxviruses | New Zealand | ||
| P00202402600 | Synthetic Chimeric Poxviruses | Indonesia | ||
| 113144133 | Synthetic Chimeric Poxviruses | Taiwan |
Synthetic Vaccinia Virus
| Application No. | Title | Country / Region | ||
| 2019/37492 | Synthetic Chimeric Vaccinia Virus | Gulf Cooperation Council | ||
| 2019/41458 | Synthetic Chimeric Vaccinia Virus | Gulf Cooperation Council | ||
| 20190101165 | Synthetic Chimeric Vaccinia Virus | Argentina | ||
| 108115290 | Synthetic Chimeric Vaccinia Virus | Taiwan R.O.C. | ||
| BR112020022181-3 | Synthetic Chimeric Vaccinia Virus | Brazil | ||
| 3099330 | Synthetic Chimeric Vaccinia Virus | Canada | ||
| 202510639606.8 | Synthetic Chimeric Vaccinia Virus | China | ||
| 19796145.1 | Synthetic Chimeric Vaccinia Virus | European Patent Office | ||
| 202017052398 | Synthetic Chimeric Vaccinia Virus | India | ||
| P00202008694 | Synthetic Chimeric Vaccinia Virus | Indonesia | ||
| 278419 | Synthetic Chimeric Vaccinia Virus | Israel | ||
| 2024-28557 | Synthetic Chimeric Vaccinia Virus | Japan | ||
| 2025-151400 | Synthetic Chimeric Vaccinia Virus | Japan | ||
| 768999 | Synthetic Chimeric Vaccinia Virus | New Zealand | ||
| 10202401171Y | Synthetic Chimeric Vaccinia Virus | Singapore | ||
| 2020/06350 | Synthetic Chimeric Vaccinia Virus | South Africa | ||
| 62021038254.0 | Synthetic Chimeric Vaccinia Virus | Hong Kong | ||
| PI 2024003859 | Synthetic Chimeric Vaccinia Virus | Malaysia | ||
| 810669 | Synthetic Chimeric Vaccinia Virus | New Zealand |
45
Poxvirus vaccine against COVID-19
| Application No. | Title | Country / Region | ||
| 17/187,678 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | U.S.A. | ||
| 114138879 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | Taiwan | ||
| 20210100512 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | Argentina | ||
| 1202200348 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | African Intellectual Property Organization | ||
| AP/P/2022/014318 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | African Regional Intellectual Property Organization | ||
| 2021226592 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | Australia | ||
| BR112022016992-2 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | Brazil | ||
| 3173996 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | Canada | ||
| 202180027983.6 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | China | ||
| 202292431 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | Eurasian Patent Office | ||
| 21715007.7 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | European Patent Office | ||
| 202217053476 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | India | ||
| P00202210244 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | Indonesia | ||
| 295925 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | Israel | ||
| 2022-551297 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | Japan | ||
| 2026-0034131 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | Japan | ||
| PI 2022004613 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | Malaysia | ||
| MX/a/2022/010588 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | Mexico | ||
| 791924 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | New Zealand | ||
| 830553 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | New Zealand | ||
| 10-2022-7033014 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | Republic of Korea | ||
| 522440323 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | Saudi Arabia | ||
| 2022/09895 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | South Africa | ||
| 523451920 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | Saudi Arabia | ||
| 62023075022.1 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | Hong Kong | ||
| 62023083678.0 | Recombinant Poxvirus Based Vaccine against SARS-CoV-2 virus | Hong Kong |
TFF2 therapeutics
| Application No. | Title | Country / Region | ||
| 17/638,761 | Modified TFF2 polypeptides | U.S.A. | ||
| 2020338947 | Modified TFF2 polypeptides | Australia | ||
| 3152665 | Modified TFF2 polypeptides | Canada | ||
| 202080071768.1 | Modified TFF2 polypeptides | China | ||
| 20781063.1 | Modified TFF2 polypeptides | European Patent Office | ||
| 202217016249 | Modified TFF2 polypeptides | India | ||
| 290910 | Modified TFF2 polypeptides Containing Compositions and Methods of Use Thereof | Israel | ||
| 2022-513154 | Modified TFF2 polypeptides | Japan | ||
| 2025-86620 | Modified TFF2 polypeptides | Japan | ||
| MX/a/2022/002337 | Modified TFF2 polypeptides | Mexico | ||
| 786004 | Modified TFF2 polypeptides | New Zealand | ||
| 824645 | Modified TFF2 polypeptides | New Zealand | ||
| 2022/03355 | Modified TFF2 polypeptides | South Africa | ||
| 62023066535.3 | Modified TFF2 polypeptides | Hong Kong | ||
| 62023066928.0 | Modified TFF2 polypeptides | Hong Kong |
Triptan Compound – Formulations
| Application No. | Title | Country / Region | ||
| 18/782,922 | Formulations Comprising Triptan Compounds | U.S.A. |
Triptan Compound – Migraine
| Application No. | Title | Country / Region | ||
| 18/797,812 | Methods of Treating Migraine | U.S.A. | ||
| 18/783,734 | Pharmaceutical Composition for Treating Migraine | U.S.A. |
46
Tianeptine – Conditions Associated with Peroxisome Proliferator-Activated Receptor
| Application No. | Title | Country / Region | ||
| 19/469,537 | (S)-Tianeptine and Use in Treating Disorders and Conditions Associated with Peroxisome Proliferator-Activated Receptor | U.S.A. | ||
| 2024245246 | (S)-Tianeptine and Use in Treating Disorders and Conditions Associated with Peroxisome Proliferator-Activated Receptor | Australia | ||
| BR112025020808-0 | (S)-Tianeptine and Use in Treating Disorders and Conditions Associated with Peroxisome Proliferator-Activated Receptor | Brazil | ||
| 3287412 | (S)-Tianeptine and Use in Treating Disorders and Conditions Associated with Peroxisome Proliferator-Activated Receptor | Canada | ||
| 202480032955.7 | (S)-Tianeptine and Use in Treating Disorders and Conditions Associated with Peroxisome Proliferator-Activated Receptor | China | ||
| 24719842.7 | (S)-Tianeptine and Use in Treating Disorders and Conditions Associated with Peroxisome Proliferator-Activated Receptor | European Patent Office | ||
| 323504 | (S)-Tianeptine and Use in Treating Disorders and Conditions Associated with Peroxisome Proliferator-Activated Receptor | Israel | ||
| 202517102234 | (S)-Tianeptine and Use in Treating Disorders and Conditions Associated with Peroxisome Proliferator-Activated Receptor | India | ||
| 2025-556587 | (S)-Tianeptine and Use in Treating Disorders and Conditions Associated with Peroxisome Proliferator-Activated Receptor | Japan | ||
| PI 2025005867 | (S)-Tianeptine and Use in Treating Disorders and Conditions Associated with Peroxisome Proliferator-Activated Receptor | Malaysia | ||
| MX/a/2025/011371 | (S)-Tianeptine and Use in Treating Disorders and Conditions Associated with Peroxisome Proliferator-Activated Receptor | Mexico | ||
| 826013 | (S)-Tianeptine and Use in Treating Disorders and Conditions Associated with Peroxisome Proliferator-Activated Receptor | New Zealand | ||
| 1120257039 | (S)-Tianeptine and Use in Treating Disorders and Conditions Associated with Peroxisome Proliferator-Activated Receptor | Saudi Arabia | ||
| 11202506430U | (S)-Tianeptine and Use in Treating Disorders and Conditions Associated with Peroxisome Proliferator-Activated Receptor | Singapore | ||
| 2025/08940 | (S)-Tianeptine and Use in Treating Disorders and Conditions Associated with Peroxisome Proliferator-Activated Receptor | South Africa |
Trademarks and Service Marks
Tonix Pharmaceuticals, Inc.
We seek trademark and service mark protection in the United States and outside of the United States where available and when appropriate. We are the owner of the following U.S. federally registered mark: TONIX PHARMACEUTICALS (Reg. No. 4656463, issued December 16, 2014).
We are the owner of the following marks for which applications for U.S. federal registration are currently pending: FYMRALIN (Serial No. 98/327953, filed December 22, 2023), MODALTIN (Serial No. 99/562722, filed December 23, 2025), RAPONTIS (Serial No. 90/562733, filed December 23, 2025), PROTECTIC (Serial No. 99/562746, filed December 23, 2025), TONIX PHARMACEUTICALS (Serial No. 98/577945, filed May 31, 2024), ANGSTRO-TECHNOLOGY (Serial No. 98/006538, filed May 22, 2023) ).
Tonix Medicines, Inc.
We are the owner of the following U.S. federally registered marks: NOTIME4MIGRAINES (Reg. No. 5392512, issued January 30, 2018); SYMTOUCH (Reg. No. 5186988, issued April 18, 2017); TOSYMRA (Reg. No. 5981221, issued February 11, 2020); TOSYMRA & DROPLET Design (Reg. No. 6124333, issued August 11, 2020); ZEMBRACE (Reg. No. 5186989, issued April 18, 2017); ZEMBRACE SYMTOUCH (Reg. No.5478282, issued May 29, 2018); DROPLET Design (Reg. No. 6117797, issued August 4, 2020); TONIX ONE LOGO Design (Serial No. 99/057051, filed February 26, 2025). We are the owner of the following International Registrations under the Madrid Protocol: TOSYMRA (Reg. No. 1501060, issued October 16, 2019 – Extensions of Protection to: Canada, European Union, Japan, Republic of Korea); ZEMBRACE (Reg. No. 1683288, issued August 17, 2022 – Extensions of Protection to: Canada, China, European Union, Israel, Mexico, United Kingdom); DROPLET Design (Reg. No. 1545038, issued June 29, 2020 – Extensions of Protection to: Canada, European Union, Israel, Japan, Norway, Republic of Korea, Switzerland, Turkey, United Kingdom); European Union: TONIX ONE LOGO Design (Registration No. 019151272, issued July 9, 2025); United Kingdom: TOSYMRA, Reg. No. UK00801501060.
47
Tonix Pharma Limited
We are the owner of the mark TONMYA for which U.S. Application Serial Number: 97/185424, filed December 22, 2021, is currently pending.
Research and Development
We have approximately 70 employees dedicated to research and development. Our research and development operations are located in Berkeley Heights, NJ, Frederick, Maryland, Dublin, Ireland and Montreal, Canada. We have used, and expect to continue to use, third parties to conduct our nonclinical and clinical studies.
Manufacturing
We have contracted with third-party cGMP-compliant contract manufacturer organizations(“CMOs”), for the manufacture of TNX-102 SL drug substance and drug products for clinical and commercial supply. Our manufacturing operations are managed and controlled out of our Dublin, Ireland offices.
All of our small molecules drug candidates are synthesized using industry standard processes, and our drug products are formulated using commercially available pharmaceutical grade excipients.
We own a 45,000 square foot facility in Massachusetts, to house our new Advanced Development Center for accelerated development and manufacturing of vaccines and biologics. This facility is currently decommissioned. It was designed for the manufacture of nonclinical and clinical investigational products for our Infectious Disease portfolio. The current focus of which is TNX-801 a Smallpox and Mpox Preventing Vaccine. This facility was decommissioned in 2024 but may be reactivated the earlier of 2027 or in the case of a national or international emergency.
Our other marketed products, Zembrace and Tosymra, are manufactured at cGMP compliant, FDA audited, U.S. based CMOs with expertise in pre-filled syringe and inhalation expertise.
Government Regulations
Government authorities in the United States, at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of products such as those we are developing.
U.S. Government Regulation
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA, and its implementing regulations, and biologics under the FDCA and the Public Health Service Act, or PHSA, and its implementing regulations. FDA approval is required before any new unapproved drug or dosage form, including a new use of a previously approved drug, can be marketed in the United States. Drugs and biologics are also subject to other federal, state and local statutes and regulations. If we fail to comply with applicable FDA or other requirements at any time during the drug development process, clinical testing, the approval process or after approval, we may become subject to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, license suspension or revocation, withdrawal of an approval, warning letters, product recalls, product seizures, placement on Import Alerts, debarment of personnel, employees or officers, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution.
The process required by the FDA before product candidates may be marketed in the United States generally involves the following:
| ● | completion of extensive preclinical laboratory tests, preclinical animal studies, and toxicity data, all performed in accordance with the good laboratory practices, or GLP, regulations; |
| ● | submission to the FDA of an IND, which must become effective before human clinical studies may begin; |
| ● | approval by an independent IRB or ethics committee representing each clinical site before each clinical study may be initiated; |
| ● | performance of adequate and well-controlled human clinical studies to establish the safety and efficacy, or in the case of a biologic, the safety, purity and potency, of the drug candidate for each proposed indication; |
| ● | preparation of and submission to the FDA of a new drug application, or NDA, or biologics license application, or BLA, which must include data from required pre-clinical studies and all pivotal clinical studies and information showing that the product can be manufactured in a controlled manner; |
| ● | a determination by the FDA within 60 days of its receipt of an NDA or BLA to file the application for review; |
| ● | review of the product application by an FDA advisory committee, where appropriate and if applicable; |
48
| ● | satisfactory completion of an FDA pre-approval inspection of the manufacturing facilities where the drug candidate is produced to assess compliance with cGMP; and |
| ● | FDA review and approval of an NDA or BLA prior to any commercial marketing or sale of the drug or biologic. |
An IND is a request for authorization from the FDA to administer an investigational new drug product to humans. The central focus of an IND submission is on the general investigational plan and the protocol(s) for human studies. The IND also includes results of animal and in vitro studies assessing the toxicology, pharmacokinetics, pharmacology and pharmacodynamic characteristics of the product; chemistry, manufacturing and controls information; and any available human data or literature to support the use of the investigational new drug. An IND must become effective before human clinical studies may begin. An IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to the proposed clinical studies. In such a case, the IND may be placed on clinical hold and the IND sponsor and the FDA must resolve any outstanding concerns or questions before clinical studies can begin. Accordingly, submission of an IND may or may not result in the FDA allowing clinical studies to commence.
Clinical Studies
Clinical studies involve the administration of the investigational new drug to human subjects under the supervision of qualified investigators in accordance with GCPs, which include the requirement that all research subjects provide their informed consent for their participation in any clinical study. Clinical studies are conducted under protocols detailing, among other things, the objectives of the study, and the parameters to be used in monitoring safety and the efficacy criteria to be evaluated. A protocol for each clinical study and any subsequent protocol amendments must be submitted to the FDA as part of the IND. Additionally, approval must also be obtained from each clinical study site’s IRB before the studies may be initiated, and the IRB must monitor the study until completed. There are also requirements governing the reporting of ongoing clinical studies and clinical study results to public registries, such as ClinicalTrials.gov.
The clinical investigation of a drug or biologic is generally divided into three or four phases. Although the phases are usually conducted sequentially, they may overlap or be combined.
| ● | Phase 1. The drug or biologic is initially introduced into healthy human subjects or patients with the target disease or condition. These studies are designed to evaluate the safety, dosage tolerance, metabolism and pharmacologic actions of the investigational new drug in humans, the side effects associated with increasing doses, and if possible, to gain early evidence on effectiveness. |
| ● | Phase 2. The drug or biologic is administered to a limited patient population to evaluate dosage tolerance and optimal dosage, identify possible adverse side effects and safety risks and preliminarily evaluate efficacy. |
| ● | Phase 3. The drug or biologic is administered to an expanded patient population, generally at geographically dispersed clinical study sites to generate enough data to statistically evaluate dosage, clinical effectiveness and safety, to establish the overall benefit-risk relationship of the investigational product and to provide an adequate basis for product approval. |
| ● | Phase 4. In some cases, the FDA may condition approval of an NDA or BLA for a drug candidate on the sponsor’s agreement to conduct additional clinical studies after approval. In other cases, a sponsor may commit to conducting or voluntarily conduct additional clinical studies after approval to gain more information about the drug. Such post-approval studies are typically referred to as Phase 4 clinical studies. |
A confirmatory or pivotal study is a clinical study that adequately meets regulatory agency requirements for the evaluation of a drug candidate’s efficacy and safety such that it can be used to justify the approval of the product. Generally, pivotal studies are Phase 3 studies, but the FDA may accept results from Phase 2 studies if the study design provides a well-controlled and reliable assessment of clinical benefit, particularly in situations where there is an unmet medical need and the results are sufficiently robust. In such cases, FDA may require post-market studies for safety and efficacy to be conducted for the drug candidate. The FDA may withdraw the approval if the results indicate that the approved drug is not safe or effective.
The FDA, the IRB or the clinical study sponsor may suspend or terminate a clinical study at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Additionally, some clinical studies are overseen by an independent group of qualified experts organized by the clinical study sponsor, known as a data safety monitoring board or committee. This group provides authorization for whether or not a study may move forward at designated check points based on access to certain data from the study. We may also suspend or terminate a clinical study based on evolving business objectives and/or competitive climate.
49
Chemistry, Manufacturing and Control Information
Companies seeking FDA approval of drugs must also develop data and information about the physical characteristics of the components of a product as well as finalize processes for manufacturing the components in commercial quantities in accordance with GMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the sponsor must develop methods for testing the identity, strength, quality, potency and purity of the final product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the components of a product candidate do not undergo unacceptable deterioration over their shelf life.
Submission of an NDA or BLA to the FDA
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, detailed investigational new drug product information is submitted to the FDA in the form of an NDA or BLA requesting approval to market the product for one or more indications. Under federal law, the submission of most NDAs and BLAs is subject to a substantial application user fee. Applications for orphan drug products are exempted from the NDA and BLA application user fees.
An NDA or BLA must include all relevant data available from pertinent preclinical and clinical studies, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls and proposed labeling, among other things. Data can come from company-sponsored clinical studies intended to test the safety and effectiveness of a use of a product, or from a number of alternative sources, including studies initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and effectiveness of the investigational product to the satisfaction of the FDA.
Once an NDA or BLA has been submitted, the FDA’s goal is to review the application within ten months after it accepts the application for filing, or, if the application relates to an unmet medical need in a serious or life-threatening indication, six months after the FDA accepts the application for filing. The review process is often significantly extended by FDA requests for additional information or clarification.
Before approving an NDA or BLA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA or BLA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP.
The FDA is required to refer an application for an investigational drug or biologic to an advisory committee or explain why such referral was not made. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the investigational product application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions and typically follows such recommendations.
The FDA’s Decision on an NDA or BLA
After the FDA evaluates the NDA or BLA and conducts inspections of manufacturing facilities, it may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the drug or biologic with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete, and the application is not ready for approval. A Complete Response Letter may require additional clinical data and/or an additional pivotal Phase 3 clinical study(ies), and/or other significant, expensive and time-consuming requirements related to clinical studies, preclinical studies or manufacturing. Even if such additional information is submitted, the FDA may ultimately decide that the NDA or BLA does not satisfy the criteria for approval. The FDA could also approve the NDA or BLA with a REMS to mitigate risks, which could include medication guides, physician communication plans or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA also may condition approval on, among other things, changes to proposed labeling, development of adequate controls and specifications or a commitment to conduct one or more post-market studies or clinical studies. Such post-market testing may include Phase 4 clinical studies and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization. The FDA may have the authority to withdraw its approval if post-market testing fails to verify the approved drug’s clinical benefit, if the applicant does not perform the required testing with due diligence, or if the any other evidence demonstrates the approved drug is not safe or effective, among other reasons. Also, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our products under development.
50
Expedited Review and Accelerated Approval Programs
The FDA has various programs, including fast track designation, breakthrough therapy designation, accelerated approval, regenerative medicine advanced therapy and priority review, that are intended to expedite the development and approval of new drugs and biologics that address unmet medical needs in the treatment of serious or life-threatening diseases and conditions. To be eligible for a fast-track designation, the FDA must determine, based on the request of a sponsor, that a product is intended to treat a serious or life-threatening disease or condition and demonstrates the potential to address an unmet medical need. The FDA may review sections of the NDA for a fast-track product on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the NDA, the FDA agrees to accept sections of the NDA and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the NDA.
The FDA may give a priority review designation to drugs that offer major advances in treatment or provide a treatment where no adequate therapy exists. A priority review means that the goal for the FDA to review an application is six months, rather than the standard review of ten months under current. These six and ten-month review periods are measured from the “filing” date rather than the receipt date for NDAs for new molecular entities, which typically adds approximately two months to the timeline for review and decision from the date of submission. Most products that are eligible for fast-track designation are also likely to be considered appropriate to receive a priority review.
In addition, products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may be eligible for accelerated approval and may be approved on the basis of adequate and well-controlled clinical studies establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require a sponsor of a drug receiving accelerated approval to perform post-marketing studies to verify and describe the predicted effect on irreversible morbidity or mortality or other clinical endpoint, and the drug may be subject to accelerated withdrawal procedures.
Moreover, under the provisions of the FDA Safety and Innovation Act passed in July 2012, a sponsor can request designation of a drug candidate as a “breakthrough therapy.” A breakthrough therapy is defined as a drug or biologic that is intended, alone or in combination with one or more other drugs or biologics, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Drugs designated as breakthrough therapies are also eligible for the other expedited review and approval programs, including accelerated approval, priority review, regenerative medicine advanced therapy, and fast-track designation. The FDA must take certain actions, such as holding timely meetings and providing advice, intended to expedite the development and review of an application for approval of a breakthrough therapy.
In addition, the 21st Century Cures Act in 2016 made the Regenerative Medicine Advanced Therapy, or RMAT, designation available for investigational drugs that are intended to treat, modify, reverse, or cure a serious condition, with preliminary clinical evidence indicating that the drug has the potential for addressing unmet medical needs for such condition. The RMAT designation is available for cell therapy, therapeutic tissue engineering products, human cell and tissue products, and combination products that use such therapies or products. The advantages of RMAT designation include those of breakthrough and fast track designations, such as early interactions with FDA and rolling review of applications, and the drug candidate with the RMAT designation may be eligible for accelerated approval. Requests for RMAT designations should be made with the IND application (if preliminary clinical evidence is available), but no later than the end-of-phase-2 meeting.
Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened.
Post-Approval Requirements
Drugs and biologics marketed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims are subject to prior FDA review and approval. There also are continuing, annual user fee requirements.
51
Manufacturers are subject to periodic unannounced inspections by the FDA and state agencies for compliance with cGMP requirements. Changes to the manufacturing process are strictly regulated and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon us and any third-party manufacturers that we may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
Discovery of previously unknown problems with a product or the failure to comply with applicable requirements may result in restrictions on a product, manufacturer or holder of an approved NDA or BLA, including withdrawal or recall of the product from the market or other voluntary, FDA-initiated or judicial action that could delay or prohibit further marketing. Also, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our products under development.
The FDA may withdraw approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical studies to assess new safety risks; or imposition of distribution restrictions or other restrictions under a REMS program. Other potential consequences include, among other things.
| ● | restrictions on the marketing or manufacturing of the product; |
| ● | complete withdrawal of the product from the market or product recalls; |
| ● | fines, warning letters or holds on post-approval clinical studies; |
| ● | refusal of the FDA to approve pending NDAs or BLAs or supplements to approved NDAs or BLAs, or suspension or revocation of product licenses or approvals; |
| ● | product seizure or detention, or refusal to permit the import or export of products; or |
| ● | injunctions or the imposition of civil or criminal penalties. |
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability.
Pediatric Exclusivity
Pediatric exclusivity is another type of non-patent marketing exclusivity in the United States and, if granted, provides for the attachment of an additional six months to the term of any existing patent or regulatory exclusivity for drug products. This six-month exclusivity may be granted if an NDA sponsor submits pediatric data that fairly respond to a written request from the FDA for such data. The data do not need to show the product to be effective in the pediatric population studied; rather, if the clinical trial is deemed to fairly respond to the FDA’s request, the additional protection is granted. If reports of requested pediatric studies are submitted to and accepted by the FDA within the statutory time limits, whatever statutory or regulatory periods of exclusivity or patent protection cover the product are extended by six months. This is not a patent term extension, but effectively extends the regulatory period during which the FDA cannot approve another application. With regard to patents, the six-month pediatric exclusivity period will not attach to any patents for which a generic (ANDA or 505(b)(2) NDA) sponsor submitted a Paragraph IV certification, unless the NDA sponsor or patent owner first obtains a court determination that the patent is valid and infringed by a proposed generic product.
Orphan Designation and Exclusivity
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biologic intended to treat a rare disease or condition, defined as a disease or condition with a patient population of fewer than 200,000 individuals in the United States, or a patient population greater than 200,000 individuals in the United States and when there is no reasonable expectation that the cost of developing and making available the drug or biologic in the United States will be recovered from sales in the United States for that drug or biologic. Orphan drug designation must be requested before submitting a BLA or NDA. After the FDA grants orphan drug designation, the generic identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA.
52
If a product that has orphan drug designation subsequently receives the first FDA approval for a particular active ingredient for the disease for which it has such designation, the product is entitled to orphan product marketing exclusivity, which means that the FDA may not approve any other applications, including a full BLA, to market the same biologic for the same use or indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity or if FDA finds that the holder of the orphan drug exclusivity has not shown that it can assure the availability of sufficient quantities of the orphan drug to meet the needs of patients with the disease or condition for which the drug was designated. Orphan drug exclusivity does not prevent the FDA from approving a different drug or biologic for the same disease or condition, or the same drug or biologic for a different disease or condition. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the BLA or NDA application user fee.
A designated orphan drug may not receive orphan drug exclusivity if it is approved for a use that is broader than the indication for which it received orphan designation. In addition, orphan drug exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or, as noted above, if the second applicant demonstrates that its product is clinically superior to the approved product with orphan exclusivity or the manufacturer of the approved product is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition.
Biosimilars and Exclusivity
The Affordable Care Act, signed into law in 2010, includes a subtitle called the Biologics Price Competition and Innovation Act of 2009, or BPCIA, which created an abbreviated approval pathway for biological products that are biosimilar to or interchangeable with an FDA-licensed reference biological product. To date, only a handful of biosimilars have been licensed under the BPCIA, although numerous biosimilars have been approved in Europe. The FDA has issued several guidance documents outlining an approach to review and approval of biosimilars.
Biosimilarity, which requires that there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity and potency, can be shown through analytical studies, human PK and PD studies, clinical immunogenicity assessments, animal studies and a clinical study or studies. Interchangeability requires that a product is biosimilar to the reference product and the product must demonstrate that it can be expected to produce the same clinical results as the reference product in any given patient and, for products that are administered multiple times to an individual, the biologic and the reference biologic may be alternated or switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. However, complexities associated with the larger, and often more complex, structures of biological products, as well as the processes by which such products are manufactured, pose significant hurdles to implementation of the abbreviated approval pathway that are still being worked out by the FDA.
Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date that the reference product was first licensed by the FDA. In addition, the approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. During this 12-year period of exclusivity, another company may still market a competing version of the reference product if the FDA approves a full BLA for the competing product containing that applicant’s own preclinical data and data from adequate and well-controlled clinical studies to demonstrate the safety, purity and potency of its product. The BPCIA also created certain exclusivity periods for biosimilars approved as interchangeable products. At this juncture, it is unclear whether products deemed “interchangeable” by the FDA will, in fact, be readily substituted by pharmacies, which are governed by state pharmacy law.
A biological product can also obtain pediatric market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric study in accordance with an FDA-issued “Written Request” for such a study.
The BPCIA is complex and continues to be interpreted and implemented by the FDA. In addition, recent government proposals have sought to reduce the 12-year reference product exclusivity period. Other aspects of the BPCIA, some of which may impact the BPCIA exclusivity provisions, have also been the subject of recent litigation. As a result, the ultimate impact, implementation, and impact of the BPCIA is subject to significant uncertainty.
Hatch-Waxman Amendments, 505(b)(2) NDAs and Exclusivity
Section 505 of the FDCA describes three types of marketing applications that may be submitted to the FDA to request marketing authorization for a new drug. A Section 505(b)(1) NDA is an application that contains full reports of investigations of safety and efficacy. A 505(b)(2) NDA is an application that contains full reports of investigations of safety and efficacy but where at least some of the information required for approval comes from investigations that were not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use from the person by or for whom the investigations were conducted. This regulatory pathway enables the applicant to rely, in part, on the FDA’s prior findings of safety and efficacy for an existing product, or published literature, in support of its application. Section 505(j) establishes an abbreviated approval process for a generic version of approved drug products through the submission of an ANDA. An ANDA provides for marketing of a generic drug product that has the same active ingredients, dosage form, strength, route of administration, labeling, performance characteristics and intended use, among other things, to a previously approved product. ANDAs are termed “abbreviated” because they are generally not required to include preclinical (animal) and clinical (human) data to establish safety and efficacy. Instead, generic applicants must scientifically demonstrate that their product is bioequivalent to, or performs in the same manner as, the innovator drug through in vitro, in vivo or other testing. The generic version must deliver the same amount of active ingredients into a subject’s bloodstream in the same amount of time as the innovator drug and can often be substituted by pharmacists under prescriptions written for the reference listed drug. In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent with claims that cover the applicant’s drug or a method of using the drug. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential competitors in support of approval of an ANDA or 505(b)(2) NDA.
53
Upon submission of an ANDA or a 505(b)(2) NDA, an applicant must certify to the FDA that (1) no patent information on the drug product that is the subject of the application has been submitted to the FDA; (2) such patent has expired; (3) the date on which such patent expires; or (4) such patent is invalid or will not be infringed upon by the manufacture, use or sale of the drug product for which the application is submitted. Generally, the ANDA or 505(b)(2) NDA cannot be approved until all listed patents have expired, except where the ANDA or 505(b)(2) NDA applicant challenges a listed patent through the last type of certification, also known as a paragraph IV certification. If the applicant does not challenge the listed patents or indicates that it is not seeking approval of a patented method of use, the ANDA or 505(b)(2) NDA application will not be approved until all of the listed patents claiming the referenced product have expired.
If the ANDA or 505(b)(2) NDA applicant has provided a Paragraph IV certification to the FDA, the applicant must send notice of the Paragraph IV certification to the NDA and patent holders once the application has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the paragraph IV certification. If the paragraph IV certification is challenged by an NDA holder or the patent owner(s) asserts a patent challenge to the paragraph IV certification, the FDA may not approve that application until the earlier of 30 months from the receipt of the notice of the paragraph IV certification, the expiration of the patent, when the infringement case concerning each such patent was favorably decided in the applicant’s favor or settled, or such shorter or longer period as may be ordered by a court. This prohibition is generally referred to as the 30-month stay. In instances where an ANDA or 505(b)(2) NDA applicant files a paragraph IV certification, the NDA holder or patent owner(s) regularly take action to trigger the 30-month stay, recognizing that the related patent litigation may take many months or years to resolve.
The FDA also cannot approve an ANDA or 505(b)(2) application until all applicable non-patent exclusivities listed in the Orange Book for the branded reference drug have expired. For example, a pharmaceutical manufacturer may obtain five years of non-patent exclusivity upon NDA approval of a new chemical entity, or NCE, which is a drug containing an active moiety that has not been approved by FDA in any other NDA. An “active moiety” is defined as the molecule responsible for the drug substance’s physiological or pharmacologic action. During that five-year exclusivity period, the FDA cannot accept for filing (and therefore cannot approve) any ANDA seeking approval of a generic version of that drug or any 505(b)(2) NDA that relies on the FDA’s approval of the drug, provided that that the FDA may accept an ANDA four years into the NCE exclusivity period if the ANDA applicant also files a Paragraph IV certification.
A drug, including one approved under Section 505(b)(2), may obtain a three-year period of exclusivity for a particular condition of approval, or change to a marketed product, such as a new formulation for a previously approved product, if one or more new clinical studies (other than bioavailability or bioequivalence studies) was essential to the approval of the application and was conducted/sponsored by the applicant. Should this occur, the FDA would be precluded from approving any ANDA or 505(b)(2) application for the protected modification until after that three-year exclusivity period has run. However, unlike NCE exclusivity, the FDA can accept an application and begin the review process during the exclusivity period.
A Section 505(b)(2) NDA may be submitted for a drug for which one or more of the investigations relied upon by the applicant was not conducted by or for the applicant and for which the applicant has no right of reference from the person by or for whom the investigations were conducted. A Section 505(b)(2) NDA may be submitted based in whole or in part on published literature or on the FDA’s finding of safety and efficacy of one or more previously approved drugs, which are known as reference drugs. Thus, the filing of a Section 505(b)(2) NDA may result in approval of a drug based on fewer clinical or nonclinical studies than would be required under a full NDA. The number and size of studies that need to be conducted by the sponsor depends on the amount and quality of data pertaining to the reference drug that are publicly available, and on the similarity of and differences between the applicant’s drug and the reference drug. In some cases, extensive, time-consuming, and costly clinical and nonclinical studies may still be required for approval of a Section 505(b)(2) NDA.
54
Our drug approval strategy for our new formulations of approved chemical entities is to submit Section 505(b)(2) NDAs to the FDA. TONMYA was approved as a Section 505(b)(2) NDA and we plan to submit an 505(b)(2)for TNX-2900 for Prader Willi Syndrome. The FDA may not agree that our product candidates are approvable as Section 505(b)(2) NDAs. If the FDA determines that a Section 505(b)(2) NDA is not appropriate and that a full NDA is required, the time and financial resources required to obtain FDA approval could substantially and materially increase and be less likely to be approved. If the FDA requires a full NDA or requires more extensive testing and development for some other reason, our ability to compete with alternative products that arrive on the market more quickly than our product candidates would be adversely impacted. If reference listed products are withdrawn from the market by the FDA for a safety reason, we may not be able to reference such products to support our anticipated 505(b)(2) NDAs, and we may be required to follow the requirements of Section 505(b)(1).
Patent Term Restoration and Extension
A patent claiming a new drug product, its method of use or its method of manufacture may be eligible for a limited patent term extension under the Hatch-Waxman Act, which permits a patent restoration of up to five years for patent term lost during product development and the FDA regulatory review. The restoration period granted on a patent covering a product is typically one-half the time between the effective date of when a clinical trial involving human beings has begun and the submission date of an application for approval, plus the time between the submission date of an application and the ultimate approval date. Patent term restoration cannot be used to extend the remaining patent term past a total of 14 years from the product’s approval date. Only one patent applicable to an approved product is eligible for the extension, and the application for the extension must be submitted prior to the expiration of the patent in question. A patent that covers multiple products for which approval is sought can only be extended in connection with one of the approvals. The USPTO reviews and approves the application for any patent term extension or restoration in consultation with the FDA.
Material Threat Medical Countermeasures
In 2016, the 21st Century Cures Act, or Act, was signed into law to support ongoing biomedical innovation. One part of the Act, Section 3086, is aimed at “Encouraging Treatments for Agents that Present a National Security Threat.” The Act created a new priority review voucher program for approved “material threat medical countermeasure applications.” The Act defines such countermeasures as drug or biological products, including vaccines intended to treat biological, chemical, radiological, or nuclear agents that present a national security threat or to treat harm from a condition that may be caused by administering a drug or biological product against such an agent. The Department of Homeland Security has identified 13 such threats, including anthrax, smallpox, Ebola/Marburg, tularemia, botulinum toxin, and pandemic influenza, which includes SARS-CoV-2.
Other Healthcare Laws and Compliance Requirements
Pharmaceutical companies are subject to additional healthcare regulation and enforcement by the federal government and by authorities in the states and foreign jurisdictions in which they conduct their business. Such laws include, without limitation, state and federal anti-kickback, fraud and abuse, false claims, privacy and security and physician sunshine laws and regulations. If their operations are found to be in violation of any of such laws or any other governmental regulations that apply, they may be subject to penalties, including, without limitation, civil and criminal penalties, damages, fines, the curtailment or restructuring of operations, exclusion from participation in federal and state healthcare programs and individual imprisonment.
Coverage and Reimbursement
Sales of any product depend, in part, on the extent to which such product will be covered by third-party payors, such as federal, state and foreign government healthcare programs, commercial insurance and managed healthcare organizations and the level of reimbursement for such product by third-party payors. Decisions regarding the extent of coverage and amount of reimbursement to be provided are made on a plan-by-plan basis. These third-party payors are increasingly reducing reimbursements for medical products, drugs and services. In addition, the U.S. government, state legislatures and foreign governments have continued implementing cost-containment programs, including price controls, restrictions on coverage and reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit sales of any product. Decreases in third-party reimbursement for any product or a decision by a third-party payor not to cover a product could reduce physician usage and patient demand for the product and also have a material adverse effect on sales. Even after FDA approves a product, failure to have the product covered by third-party payors may have material adverse effect on sales. Federal and state governments continue to promulgate new policies and regulations; such policies and regulations may have material adverse effect on sales. These laws and regulations may restrict, prohibit, or preventing us from implementing a wide range of pricing, discounting, marketing, promotion, sales commission, incentive programs, and other business activities. No uniform policy of coverage and reimbursement among third-party payors exists in the United States. Such payors often rely upon Medicare coverage policy establishing their coverage and reimbursement policies. However, each payor makes independent and separate decisions regarding the extent of coverage and amount of reimbursement to be provided.
55
Legislative and Regulatory Changes, Including Health Care Reform
The laws and regulation that affect our business are subject to change from time to time, and entirely new laws and regulations are sometimes adopted. In particular, healthcare reforms that have been adopted, and that may be adopted in the future, could result in further reductions in coverage and levels of reimbursement for pharmaceutical products, increases in rebates payable under U.S. government rebate programs and additional downward pressure on pharmaceutical product prices. Individual states in the United States have also become increasingly active in implementing regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, proposing to encourage importation from other countries and bulk purchasing. It is unclear to what extent these and other statutory, regulatory, and administrative initiatives will be enacted and implemented.
Foreign Corrupt Practices Act
Our business activities may be subject to the Foreign Corrupt Practices Act, or FCPA, and similar anti-bribery or anti-corruption laws, regulations or rules of other countries in which we operate. The FCPA generally prohibits offering, promising, giving, or authorizing others to give anything of value, either directly or indirectly, to a non-U.S. government official in order to influence official action, or otherwise obtain or retain business. The FCPA also requires public companies to make and keep books and records that accurately and fairly reflect the transactions of the corporation and to devise and maintain an adequate system of internal accounting controls. Our business is heavily regulated and therefore involves significant interaction with public officials, including officials of non-U.S. governments. Additionally, in many other countries, the health care providers who prescribe pharmaceuticals are employed by their government, and the purchasers of pharmaceuticals are government entities; therefore, our dealings with these prescribers and purchasers are subject to regulation under the FCPA. There is no certainty that all of our employees, agents, suppliers, manufacturers, contractors, or collaborators, or those of our affiliates, will comply with all applicable laws and regulations, particularly given the high level of complexity of these laws. Violations of these laws and regulations could result in fines, criminal sanctions against us, our officers, or our employees, the closing down of facilities, including those of our suppliers and manufacturers, requirements to obtain export licenses, cessation of business activities in sanctioned countries, implementation of compliance programs, and prohibitions on the conduct of our business. Any such violations could include prohibitions on our ability to offer our products in one or more countries as well as difficulties in manufacturing or continuing to develop our products, and could materially damage our reputation, our brand, our international expansion efforts, our ability to attract and retain employees, and our business, prospects, operating results, and financial condition.
European Union Drug Development
In the European Union, our product candidates may also be subject to extensive regulatory requirements. As in the United States, medicinal products can only be marketed if a marketing authorization from the competent regulatory agencies has been obtained.
Similar to the United States, the various phases of preclinical and clinical research in the European Union are subject to significant regulatory controls. Clinical trials of medicinal products in the European Union must be conducted in accordance with European Union, national regulations and international standards for good clinical practice, or GCP.
Clinical trials are currently governed by EU Clinical Trials Directive 2001/20/EC that set out common rules for the control and authorization of clinical trials in the European Union.
To improve the current system, Regulation (EU) No 536/2014 on clinical trials on medicinal products for human use was adopted in 2014. The Regulation aims at harmonizing and streamlining the clinical trials authorization process, simplifying adverse event reporting procedures, improving the supervision of clinical trials, and increasing their transparency, notably via a clinical trial information system set up by the EMA. The new Regulation expressly provides that it will not be applied before six months after the publication of a notice delivered by the European Commission on the European Union clinical trial portal and database. As such notice requires a successful (partial) audit of the database and as that database is still under development, there is no scheduled application date yet. Pursuant to the transitory provisions of the new regulation, the Clinical Trials Directive 2001/20/EC will still apply for three years after the implementation of the European Union clinical trial portal and database. Thus, the sponsor has the possibility to choose between the requirements of the directive and the regulation for a period of three years from the entry into force of the regulation.
56
European Union Drug Review and Approval
In the EEA (which is comprised of the 28 Member States of the European Union plus Norway, Iceland and Liechtenstein), medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA. MAs may be granted either centrally (Community MA) or nationally (National MA).
The Community MA is issued centrally by the European Commission through the Centralized Procedure, based on the opinion of the CHMP of the EMA and is valid throughout the entire territory of the EEA. The Centralized Procedure is mandatory for certain types of products such as orphan medicinal products and medicinal products containing a new active substance indicated for the treatment of neurodegenerative disorders. The Centralized Procedure is optional for products containing a new active substance not yet authorized in the EEA, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the European Union.
National MAs are issued nationally by the competent authorities of the Member States of the EEA and only cover their respective territory. National MAs are available for products not falling within the mandatory scope of the Centralized Procedure. We do not foresee that any of our current product candidates will be suitable for a National MA as they fall within the mandatory criteria for the Centralized Procedure. Therefore, our product candidates will be approved through Community MAs.
Under the above-described procedures, before granting the MA, the EMA or the competent authorities of the Member States of the EEA make an assessment of the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy.
The pediatric use marketing authorization, or PUMA, is a dedicated marketing authorization for medicinal products indicated exclusively for use in the pediatric population, with, if necessary, an age-appropriate formulation. Pursuant to Regulation (EC) No. 1901/2006 (The “Pediatric Regulation”), all PUMA applications for marketing authorization for new medicines must include to be valid, in addition to the particulars and documents referred to in Directive 2001/83/EC, the results of all studies performed and details of all information collected in compliance with a pediatric investigation plan agreed between regulatory authorities and the applicant, unless the medicine is exempt because of a deferral or waiver of the EMA.
Before the EMA is able to begin its assessment of a Community MA application, it will validate that the applicant has complied with the agreed pediatric investigation plan. The applicant and the EMA may, where such a step is adequately justified, agree to modify a pediatric investigation plan to assist validation. Modifications are not always possible; may take longer to agree than the period of validation permits; and may still require the applicant to withdraw its marketing authorization application and to conduct additional non-clinical and clinical studies. Products that are granted a MA on the basis of the pediatric clinical trials conducted in accordance with the Pediatric Investigation Plan, or PIP, are eligible for a six-month extension of the protection under a supplementary protection certificate (if any is in effect at the time of approval) or, in the case of orphan medicinal products, a two-year extension of the orphan market exclusivity. This pediatric reward is subject to specific conditions and is not automatically available when data in compliance with the PIP are developed and submitted.
Orphan Drugs
In the European Union, Regulation (EC) No 141/2000 of the European Parliament and of the Council of December 16, 1999 on orphan medicinal products, as amended, states that a drug shall be designated as an orphan drug if its sponsor can establish that the three following cumulative conditions are met:
| ● | the product is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition; |
| ● | the prevalence of the conditions is not more than five in ten thousand persons in the European Union when the application is made, or that it is intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition in the European Union and that without incentives it is unlikely that the marketing of the drug in the European Union would generate sufficient return to justify the necessary investment; and |
| ● | that there is no satisfactory method of diagnosis, prevention or treatment of the condition in question that has been authorized in the European Union or, if such method exists, that the drug will be of significant benefit to those affected by that condition. |
Pursuant to Regulation (EC) No. 847/2000 of April 27, 2000 laying down the provisions for implementation of the criteria for designation of a medicinal product as an orphan medicinal product and definitions of the concepts “similar medicinal product” and “clinical superiority”, an application for the designation of a drug as an orphan drug must be submitted at any stage of development of the drug before filing of a MA application.
57
The European Union offers incentives to encourage the development of designated orphan medicines (protocol assistance, fee reductions, etc.) and provides opportunities for market exclusivity. Pursuant to abovementioned Regulation (EC) No. 141/2000, products receiving orphan designation in the European Union can obtain market exclusivity for a certain number of years in the European Union following the marketing approval.
If a Community MA in respect of an orphan drug is granted, regulatory authorities will not, for a period of usually ten years, accept another application for a MA, or grant a MA or accept an application to extend an existing MA, for the same therapeutic indication, in respect of a similar drug. This period may however be reduced to six years if, at the end of the fifth year, it is established, in respect of the drug concerned, that the above-mentioned criteria for orphan drug designation are no longer met, in other words, when it is shown on the basis of available evidence that the product is sufficiently profitable not to justify maintenance of market exclusivity.
Pursuant to Regulation No. 1901/2006, for orphan medicinal products, instead of an extension of the supplementary protection certificate, the ten-year period of orphan market exclusivity should be extended to 12 years if the requirement for data on use in the pediatric population is fully met (i.e. when the request contains the results of all studies carried out under the approved PIP and when the declaration attesting the conformity of the request to this PIP is included in the MA).
Notwithstanding the foregoing, a MA may be granted, for the same therapeutic indication, to a similar drug if:
| ● | the holder of the MA for the original orphan drug has given its consent to the second applicant; |
| ● | the holder of the MA for the original orphan drug is unable to supply sufficient quantities of the drug; or |
| ● | the second applicant can establish in the application that the second drug, although similar to the orphan drug already authorized, is safer, more effective or otherwise clinically superior. |
The abovementioned Regulation (EC) No. 141/2000 provides for other incentives regarding orphan medicinal products.
Post-Approval Controls
The holder of a MA must comply with EU requirements applicable to manufacturing, marketing, promotion and sale of medicinal products. In particular, the holder of the MA must establish and maintain a pharmacovigilance system and appoint an individual qualified person for pharmacovigilance, or QPPV, who is responsible for oversight of that system and who will reside and operate in the EU. Key obligations include safety expedited reporting of suspected serious adverse reactions and submission of periodic safety update reports, or PSURs.
All new MAs must include a risk management plan, or RMP, to submit to the EMA, describing the risk management system that the company will put in place and documenting measures to prevent or minimize the risks associated with the product. The regulatory authorities may also impose specific obligations as a condition of the MA. Such risk-minimization measures or post-authorization obligations may include additional safety monitoring, more frequent submission of PSURs, or the conduct of additional clinical trials or post-authorization safety studies. RMPs and PSURs are routinely available to third parties requesting access, subject to limited redactions. All advertising and promotional activities for the product must be consistent with the approved summary of product characteristics, and therefore all off-label promotion is prohibited. Direct-to-consumer advertising of prescription medicines is also prohibited in the European Union. Although general requirements for advertising and promotion of medicinal products are established under EU directives, the details are governed by regulations in each EU Member State and can differ from one country to another.
Reimbursement
The European Union provides options for its Member States to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A Member State may approve a specific price for the medicinal product, or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. For example, in France, effective market access will be supported by agreements with hospitals and products may be reimbursed by the Social Security Fund. The price of medicines covered by national health insurance is negotiated with the Economic Committee for Health Products, or CEPS. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our product candidates. Historically, products launched in the European Union do not follow price structures of the United States and generally prices tend to be significantly lower.
58
Human Capital Resources
As of March 12, 2026, we had 142 full-time employees, of whom 29 hold M.D. or Ph.D. degrees. We have 70 employees dedicated to research and development. We have 10 employees supporting sales administration and marketing initiatives; customer service requests and top-tier headache specialists. None of our employees are represented by a collective bargaining agreement. We believe that the skills, experience and industry knowledge of our key employees significantly benefit our operations and performance. Our research and development operations are located in Berkeley Heights, New Jersey, Dartmouth, Massachusetts, Frederick, Maryland, Dublin, Ireland and Montreal, Canada. We have used, and expect to continue to use, third parties to conduct our nonclinical and clinical studies as well as part-time employees.
Employee health and safety in the workplace is one of our core values.
Employee levels are managed to align with the pace of business and management believes it has sufficient human capital to operate its business successfully.
Corporate Information
We lease the space for our principal executive offices, which are located at 200 Connell Drive, Suite 3100, Berkeley Heights, New Jersey 07922, and our telephone number is (862) 799 8599. Our website address is www.tonixpharma.com. We do not incorporate the information on our websites into this Annual Report, and you should not consider such information part of this Annual Report.
We were incorporated on November 16, 2007, under the laws of the State of Nevada as Tamandare Explorations Inc. On October 11, 2011, we changed our name to Tonix Pharmaceuticals Holding Corp.
ITEM 1A – Risk Factors
Summary of Risk Factors
| ● | We have a history of operating losses and expect to incur losses for the foreseeable future. We may never achieve profitability. |
| ● | We expect a number of factors to cause our operating results to fluctuate on a quarterly and annual basis, which may make it difficult to predict our future performance. |
| ● | Our independent registered public accounting firm has included an explanatory paragraph relating to our ability to continue as a going concern in its report on our audited financial statements. |
| ● | We will need additional capital to fund our operations. |
| ● | Our prospects are highly dependent on the success of TONMYA. If we are unable to successfully commercialize or maintain approval for TONMYA, our business will be materially adversely affected. |
| ● | We have a limited operating history and only recently launched TONMYA in the United States, which may make it difficult to evaluate the prospects for our future viability. |
| ● | TONMYA and our migraine products remain subject to ongoing regulatory review, and if we fail to comply with continuing regulations, we could lose our approval and the sale of TONMYA or our migraine products could be suspended. |
| ● | If estimates of the size of the potential market for TONMYA are overstated or data we have used to identify prescribing healthcare providers is inaccurate, our ability to earn revenue to support our business could be materially adversely affected. |
| ● | Coverage and adequate reimbursement may not be available for our products. |
| ● | Healthcare legislative or regulatory reform measures, including government restrictions on pricing and reimbursement, may have a negative impact on our business and results of operations. |
| ● | Even if we obtain regulatory approval to market our product candidates, our product candidates may not be accepted by the market. |
| ● | Uncertainty in Government funding and in-kind support policies and programs may adversely affect our business. |
| ● | Our product candidates are novel and still in development. |
| ● | If preclinical and nonclinical testing or clinical studies for our product candidates are unsuccessful or delayed, we will be unable to meet our anticipated development and commercialization timelines. |
| ● | We are subject to extensive and costly government regulation. |
| ● | Our product candidates may cause serious adverse events, or SAEs, or undesirable side effects. |
| ● | Any breakthrough, fast track or orphan drug designation or grant of priority review status by the FDA may not actually lead to the benefits we anticipate. |
| ● | Even if approved, our product candidates will be subject to extensive post-approval regulation. |
| ● | Our relationships with customers, physicians and third-party payors is subject to federal and state healthcare laws and regulations |
| ● | If we obtain approval to commercialize any approved products outside of the United States, a variety of risks associated with international operations could materially adversely affect our business. |
| ● | Governments outside the United States tend to impose strict price controls, which may adversely affect our revenues, if any. |
| ● | If the FDA approves generic products that compete with TONMYA, sales of TONMYA would be adversely affected. |
| ● | The biopharmaceutical industry is subject to extensive regulatory obligations and policies that may be subject to change, including due to judicial challenges. |
| ● | Reductions in staffing and funding at FDA and other federal agencies could cause delays in the development and approval of our products. |
| ● | We may never receive regulatory approval to market our current or future product candidates outside of the U.S. |
| ● | Our future growth may depend, in part, on our ability to penetrate foreign markets, where we would be subject to additional regulatory burdens and other risks and uncertainties that could materially adversely affect our business. |
| ● | There may not be market interest in TNX-801. |
| ● | If technology developed for the purposes of developing new medicines or vaccines can be applied to the creation or development of biological weapons, then our technology may be considered “dual use” technology and be subject to limitations on public disclosure or export. |
| ● | Competition and technological change may make our product candidates and technologies less attractive or obsolete. |
| ● | Failure to protect our intellectual property could materially adversely affect our business. |
| ● | We are dependent on license relationships with third parties for certain of our drug development programs. |
| ● | We may be involved in lawsuits to protect or enforce our patents, which could be expensive and time consuming. |
| ● | If we infringe the rights of third parties we could be prevented from selling products, forced to pay damages, and defend against litigation. |
| ● | There are risks to our intellectual property based on our international business initiatives. |
| ● | We may be unable to protect the confidentiality of our trade secrets, thus harming our business and competitive position. |
59
| ● | Third parties may assert that our employees or consultants have wrongfully used or disclosed confidential information or misappropriated trade secrets. |
| ● | We may need to license intellectual property from third parties, and such licenses may not be available or may not be available on commercially reasonable terms. |
| ● | We rely on third parties to conduct, supervise and monitor our clinical studies, and if those third parties perform in an unsatisfactory manner, it may harm our business. |
| ● | We may use our financial and human resources to pursue a particular research program or product candidate and fail to capitalize on programs or product candidates that may be more profitable or for which there is a greater likelihood of success. |
| ● | We will need to expand our operations and increase the size of our company, and we may experience difficulties in managing growth. |
| ● | Our executive officers and other key personnel are critical to our business, and our future success depends on our ability to retain them. |
| ● | If we are unable to hire additional qualified personnel, our ability to grow our business may be harmed. |
| ● | We rely on, and expect to continue to rely on, third parties to manufacture our marketed products and the compounds used in our studies. |
| ● | Failure by our third-party manufacturers to comply with the regulatory guidelines could delay or prevent the completion of clinical studies, the approval of any product candidates or the commercialization of our products. |
| ● | We are exposed to cybersecurity and data privacy risks. |
| ● | Adverse global conditions, including economic uncertainty, may negatively impact our financial results. |
| ● | Our internal computer systems, or those of our CROs or other contractors or consultants, may fail or suffer security breaches. |
| ● | Corporate and academic collaborators may take actions to delay, prevent, or undermine the success of our products. |
| ● | Data provided by collaborators and others upon which we rely that has not been independently verified could turn out to be false, misleading, or incomplete. |
| ● | Our product candidates may face competition sooner than expected. |
| ● | We may be unable to establish, or engage third-parties to provide, effective marketing, sales and distribution capabilities. |
| ● | We face the risk of product liability claims and may not be able to obtain insurance. |
| ● | Potential claims relating to improper handling, storage or disposal of hazardous chemicals used in our business could affect us and be time-consuming and costly. |
| ● | Our insurance policies are expensive and protect us only from some business risks, which will leave us exposed to significant uninsured liabilities. |
| ● | If we retain collaborative partners and our partners do not satisfy their obligations, we will be unable to develop our partnered product candidates. |
| ● | We face risks in connection with existing and future collaborations with respect to the development, manufacture, and commercialization of our product candidates. |
| ● | We face risks in connection with the testing, production and storage of our vaccine product candidates. |
| ● | Changes in tax laws could adversely affect our business and financial condition. |
| ● | Sales of additional shares of our common stock could cause the price of our common stock to decline. |
| ● | We may not be able to maintain compliance with the Listing Rules of the NASDAQ Stock Exchange. |
| ● | An active trading market for our common stock may not be sustained. |
| ● | The market price of our common stock has been extremely volatile in the past and may be volatile in the future due to numerous circumstances beyond our control. |
| ● | A “short squeeze” due to a sudden increase in demand for shares of our common stock that largely could lead to extreme price volatility in shares of our common stock. |
| ● | We do not anticipate paying dividends on our common stock. |
| ● | The rights of the holders of common stock may be impaired by the potential issuance of preferred stock. |
| ● | If we fail to comply with the rules under the Sarbanes-Oxley Act of 2002 related to accounting controls and procedures, or if we discover material weaknesses and deficiencies in our internal control and accounting procedures, our stock price could decline significantly and raising capital could be more difficult. |
| ● | If securities or industry analysts do not publish research or reports about our business, or if they change their recommendations regarding our stock adversely, our stock price and trading volume could decline. |
| ● | Other companies may have difficulty acquiring us, even if doing so would benefit our stockholders, due to provisions under our corporate charter and bylaws, as well as Nevada law. |
| ● | Our bylaws designate the Eighth Judicial District Court of Clark County, Nevada as the sole and exclusive forum for certain types of actions and proceedings that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers, employees or agents. |
RISKS RELATED TO OUR FINANCIAL CONDITION AND CAPITAL REQUIREMENTS
We have a history of operating losses and expect to incur losses for the foreseeable future. We may never achieve profitability.
We started generating revenues from product sales in the third quarter of 2023, and from sales of TONMYA in the fourth quarter of 2025. We have incurred losses in each year of our operations, and we expect to continue to incur operating losses for the foreseeable future as our sales and marketing, research, development, preclinical and nonclinical testing, and clinical study activities increase, and if and when we acquire rights to additional products and product candidates. The amount of future losses and when, if ever, we will achieve profitability are uncertain. Failure to achieve profitability could diminish our ability to sustain operations, pay dividends on our common stock, obtain additional required funds and make required payments on any future indebtedness. We have three products that have generated commercial revenue in the past two years, but we do not expect revenues from the commercial sale of products to exceed expenses in the near future. Our ability to generate revenue and achieve profitability will depend on, among other things, successfully commercializing our products; establishing a favorable competitive position; successful completion of the development of our product candidates; obtaining necessary regulatory approvals from the FDA; establishing manufacturing, sales, and marketing arrangements with third parties; and raising sufficient funds to finance our activities. Many of these factors will depend on circumstances beyond our control. We might not succeed at any of these undertakings. If we are unsuccessful at some or all of these undertakings, our business, prospects, and results of operations may be materially adversely affected.
60
We expect a number of factors to cause our operating results to fluctuate on a quarterly and annual basis, which may make it difficult to predict our future performance.
Our financial condition has varied significantly in the past and will continue to fluctuate from quarter-to-quarter and year-to-year due to a variety of factors, many of which are beyond our control. Factors relating to our business that may contribute to these fluctuations include other factors described elsewhere in this Annual Report and include, among other things:
| ● | our ability to obtain additional funding to commercialize our products and develop our product candidates; |
| ● | market acceptance of our products; |
| ● | the ability of patients or healthcare providers to obtain coverage of or sufficient reimbursement for our products; |
| ● | our ability to maintain regulatory approval for TONMYA and our migraine products, and to obtain regulatory approval for our product candidates in the United States and foreign jurisdictions; |
| ● | delays in the commencement, enrollment and timing of clinical studies; |
| ● | the success of our clinical studies through all phases of clinical development; |
| ● | any delays in regulatory review and approval of product candidates in clinical development; |
| ● | potential nonclinical toxicity and/or side effects of our products or product candidates that could delay or prevent commercialization, limit the indications for any approved drug, require the establishment of a REMS, or cause an approved drug to be taken off the market; |
| ● | our ability to establish or maintain collaborations, licensing or other arrangements; |
| ● | competition from existing or new products; |
| ● | our ability to leverage our proprietary technology platform to discover and develop additional product candidates; |
| ● | our ability and our licensors’ abilities to successfully obtain, maintain, defend and enforce intellectual property rights important to our business; and |
| ● | potential product liability claims. |
Accordingly, the results of any quarterly or annual periods should not be relied upon as indications of future operating performance.
Our independent registered public accounting firm has included an explanatory paragraph relating to our ability to continue as a going concern in its report on our audited financial statements.
If we are unable to obtain sufficient funding, our business, prospects, financial condition and results of operations will be materially and adversely affected, and we may be unable to continue as a going concern. For example, we anticipate that our existing cash and cash equivalents will enable us to maintain our current operations into the first quarter of 2027. If we are unable to continue as a going concern, we may have to liquidate our assets and may receive less than the value at which those assets are carried on our consolidated financial statements, and investors will likely lose all or a part of their investment. Future reports from our independent registered public accounting firm may continue to include statements expressing substantial doubt about our ability to continue as a going concern. If we seek additional financing to fund our business activities in the future and there remains substantial doubt about our ability to continue as a going concern, investors or other financing sources may be unwilling to provide additional funding on commercially reasonable terms or at all. In connection with our management’s assessment, our report from our independent registered public accounting firm for the fiscal year ended December 31, 2025, includes an explanatory paragraph stating that our recurring losses from operations and net capital deficiency raise substantial doubt about our ability to continue as a going concern.
61
We will need additional capital to fund our operations. If additional capital is not available or is available at unattractive terms, we may be forced to delay, reduce the scope of or eliminate our research and development programs, reduce our commercialization efforts or curtail our operations.
In order to successfully commercialize our marketed products and develop and bring our product candidates to market, we must commit substantial resources to costly and time-consuming commercialization activities, research, preclinical and nonclinical testing, clinical studies and the buildout of our sales, research and development and manufacturing facilities. We anticipate that our existing cash and cash equivalents will enable us to maintain our current operations until the first quarter of 2027. We anticipate using our cash and cash equivalents to commercialize TONMYA and to fund further research and development with respect to our product candidates. We will, however, need to raise additional funding sooner if our business or operations change in a manner that consumes available resources more rapidly than we anticipate. Our requirements for additional capital will depend on many factors, including:
| ● | successful commercialization of our marketed products; |
| ● | development of marketing and sales capabilities; |
| ● | market acceptance of our products; |
| ● | the time and costs involved in obtaining regulatory approval for our product candidates; |
| ● | costs associated with protecting our intellectual property rights; and |
| ● | payments received under future collaborative agreements, if any. |
To the extent we raise additional capital through the sale of equity securities, the issuance of those securities could result in dilution to our shareholders. In addition, if we obtain debt financing, a substantial portion of our operating cash flow may be dedicated to the payment of principal and interest on such indebtedness, thus limiting funds available for our business activities. If adequate funds are not available, we may be required to delay, reduce the scope of or eliminate our research and development programs, reduce our commercialization efforts or curtail our operations. In addition, we may be required to obtain funds through arrangements with collaborative partners or others that may require us to relinquish rights to technologies, product candidates or products that we would otherwise seek to develop or commercialize ourselves or license rights to technologies, product candidates or products on terms that are less favorable to us than might otherwise be available.
We will require substantial funds to support our commercialization, research and development activities, and the anticipated costs of preclinical and nonclinical testing and clinical studies, regulatory approvals and eventual commercialization of our product candidates. Such additional sources of financing may not be available on favorable terms, if at all. If we do not succeed in raising additional funds on acceptable terms, we may be unable to commence or complete clinical studies or obtain approval of any product candidates from the FDA and other regulatory authorities. In addition, we could be forced to discontinue product development, forego sales and marketing efforts and forego attractive business opportunities. Any additional sources of financing will likely involve the issuance of our equity securities, which will have a dilutive effect on our shareholders.
There is no assurance that we will be successful in raising the additional funds needed to fund our business plan. If we are not able to raise sufficient capital in the near future, our continued operations will be in jeopardy, and we may be forced to cease operations and sell or otherwise transfer all or substantially all of our remaining assets.
RISKS RELATED TO OUR BUSINESS
Our prospects are highly dependent on the success of TONMYA. If we are unable to successfully commercialize or maintain approval for TONMYA, our business, financial condition, results of operations and prospects and the value of our common stock will be materially adversely affected.
In August 2025, the FDA granted approval of TONMYA for the treatment of fibromyalgia. We have invested, and continue to invest, significant efforts and financial resources in the launch of TONMYA. We have never, as an organization, launched any other product, and there is no guarantee that we will be able to successfully commercialize TONMYA. There are numerous examples of failures to meet high expectations of market potential, including by pharmaceutical companies with more experience and resources than us. We believe that the commercial success of TONMYA depends on many factors, including the following:
| ● | the acceptance of TONMYA by physicians, patients and third-party payors; |
| ● | our ability to effectively educate healthcare providers and patients on the risks and clinical benefits of TONMYA; |
| ● | the efficacy, cost, approved use, and side-effect profile of TONMYA relative to competitive treatment regimens for fibromyalgia; |
| ● | TONMYA may compete with the off-label use of currently marketed products and other therapies in development that may in the future obtain approval for fibromyalgia; |
| ● | the effectiveness of our commercial strategy for the marketing of TONMYA, including our pricing strategy and the effectiveness of our efforts to obtain adequate third-party reimbursement; |
62
| ● | developing, maintaining and successfully monitoring commercial manufacturing arrangements for TONMYA with third-party manufacturers to ensure they meet our standards and those of regulatory authorities, including the FDA, which extensively regulate and monitor pharmaceutical manufacturing facilities; |
| ● | our ability to negotiate and enter into any additional commercial, supply and distribution contracts to support commercialization efforts, and to hire and manage additional qualified personnel; |
| ● | our ability to meet the demand for commercial supplies of TONMYA at acceptable costs; |
| ● | our ability to remain compliant with laws and regulations that apply to us and our commercial activities; |
| ● | the actual market-size, ability to identify targeted patients and the demographics of patients eligible for TONMYA, which may be different than what we currently expect; |
| ● | the occurrence of any side effects, adverse reactions or misuse, or any unfavorable publicity in these areas; |
| ● | our ability to obtain, maintain or enforce our patents and other intellectual property rights; and |
| ● | the effect of current and future health care legislation in the United States. |
While we believe that TONMYA has a commercially competitive profile, we cannot accurately predict the amount of time needed to attain a commercially successful profile or the amount of revenue that would be generated from sales of TONMYA, and there is no guarantee that we will be able to maintain or increase product sales for TONMYA or any of our marketed products. While we have established our commercial team and hired our U.S. sales force, we will need to further expand and develop the team in order to continue to grow the business. Even if we are successful in developing our commercial team, there are many factors that could negatively impact sales of our marketed products or cause commercialization efforts to be unsuccessful, including several factors that are outside our control. If the continued commercialization of our marketed products or future sales are less successful than expected or perceived as disappointing, our stock price could decline significantly, and our long-term success company could be harmed.
We have a limited operating history and only recently launched TONMYA in the United States, which may make it difficult to evaluate the prospects for our future viability.
We are still in the relatively early stages of our transition from a clinical-stage to a commercial-stage company. Our operations to date have been primarily limited to conducting research and development activities, including preclinical studies and clinical trials and, more recently, commercializing our migraine products and launching TONMYA. We have not yet demonstrated an ability to generate significant revenues, or to conduct sales and marketing activities on a long-term sustained basis necessary for successful product commercialization. Initial sales of TONMYA may not be predictive of long-term commercial results.
We may encounter unforeseen expenses, difficulties, complications, delays and other known or unknown factors in achieving our business objectives. Accordingly, you should consider our prospects in light of the costs, uncertainties, delays and difficulties frequently encountered by companies in the early commercial stage, especially pharmaceutical companies such as ours. Any predictions about our future success or viability may not be as accurate as they could be if we had a longer operating history with these activities.
TONMYA and our migraine products remain subject to ongoing regulatory review, and if we fail to comply with continuing regulations, we could lose our approval and the sale of TONMYA or our migraine products could be suspended.
Even though we received FDA approval for TONMYA, the manufacturing, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion, sampling, and record keeping related to TONMYA and our migraine products will remain subject to extensive regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGMP regulations, and GCPs for any clinical trials that we conduct post-approval, all of which may result in significant expense and limit our ability to commercialize TONMYA and our migraine products. As such, we and our contract manufacturers will be subject to periodic review and inspections to assess compliance with cGMP and adherence to commitments made in any NDA or other marketing application and previous responses to inspection observations. For certain commercial prescription drug products, manufacturers and other parties involved in the supply chain must also meet chain of distribution requirements and build electronic, interoperable systems for product tracking and tracing and for notifying the FDA of counterfeit, diverted, stolen and intentionally adulterated products or other products that are otherwise unfit for distribution in the United States. Accordingly, we and others with whom we work must continue to expend time, money, and effort in all areas of regulatory compliance, including manufacturing, production and quality control. The FDA may also require a REMS program for TONMYA or any future product candidates, which could include requirements for a medication guide, physician communication plans or additional elements to ensure safe use, such as restricted distribution methods, patient registries and other risk minimization tools.
63
If we fail to comply with the regulatory requirements of the FDA and other applicable domestic and foreign regulatory authorities, or previously unknown problems with any of our marketed products, manufacturer, or manufacturing process are discovered, we could be subject to administrative or judicially imposed sanctions, including:
| ● | restrictions on marketing or manufacturing; | |
| ● | withdrawal of the product from the market; | |
| ● | holds on clinical trials; | |
| ● | warning letters or untitled letters; | |
| ● | civil or criminal penalties; | |
| ● | fines; | |
| ● | injunctions; | |
| ● | product seizures or detentions; | |
| ● | pressure to initiate voluntary product recalls; | |
| ● | suspension or withdrawal of regulatory approvals; and | |
| ● | refusal to approve supplements to approved applications. |
If any of these events occur, our ability to sell the affected product may be impaired, and we may incur substantial additional expense to comply with regulatory requirements, which could adversely affect our business, financial condition and results of operations.
If estimates of the size of the potential market for TONMYA are overstated or data we have used to identify prescribing healthcare providers is inaccurate, our ability to earn revenue to support our business could be materially adversely affected.
We have relied on external sources, including market research funded by us and third parties, and internal analyses and calculations to estimate the potential market opportunities for TONMYA. The externally sourced information used to develop these estimates has been obtained from sources we believe to be reliable, but we have not verified the data from such sources, and their accuracy and completeness cannot be assured. With respect to TONMYA, our internal analyses and calculations are based upon management’s understanding and assessment of numerous inputs and market conditions. These understandings and assessments necessarily require assumptions subject to significant judgment and may prove to be inaccurate. As a result, our estimates of the size of these potential market for TONMYA could prove to be overstated, perhaps materially.
In addition, we are relying on third-party data to identify the prescribers who treat the majority of fibromyalgia patients in the United States; however, we may not be marketing to the appropriate prescribers and may therefore be limiting our market opportunity.
Coverage and adequate reimbursement may not be available for our products, which could make it difficult for us to sell our products profitably.
Market acceptance and sales of our products depends in part on the extent to which reimbursement for these drugs and related treatments will be available from third-party payors, including government health administration authorities, pharmacy benefit managers (“PBMs”), managed care organizations and other private health insurers. Third-party payors decide which therapies they will pay for and establish reimbursement levels. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own coverage and reimbursement policies. However, decisions regarding the extent of coverage and amount of reimbursement to be provided for any drug is made on a payor-by-payor basis. If PBMs other payers or payer affiliates deny formulary inclusion, impose unfavorable tiering, or require burdensome utilization management restrictions, patient access to our products could be significantly limited. Moreover, one payor’s determination to provide coverage for a drug does not assure that other payors will also provide coverage, and adequate reimbursement, for the drug. Additionally, a third-party payor’s decision to provide coverage for a therapy does not imply that an adequate reimbursement rate will be approved. Each payor determines whether or not it will provide coverage for a therapy, what amount it will pay the manufacturer for the therapy, and on what tier of its formulary it will be placed. The position on a payor’s list of covered drugs, or formulary, generally determines the co-payment that a patient will need to make to obtain the therapy and can strongly influence the adoption of such therapy by patients and physicians. Patients who are prescribed treatments for their conditions and providers prescribing such services generally rely on third-party payors to reimburse all or part of the associated healthcare costs. Patients are unlikely to use our drugs unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of our drugs.
A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. We cannot be sure that coverage and reimbursement will be available or continue to be available for any drug that we commercialize and, if reimbursement is available, what the level of reimbursement will be. Inadequate coverage and reimbursement may impact the demand for, or the price of, any drug we commercialize. If coverage and adequate reimbursement are not available, or are available only to limited levels, we may not be able to successfully commercialize our current and any future drug products.
64
Access to prescription drugs in the U.S. commercial market is largely controlled by three dominant entities: Emisar/Optum/United HealthCare, Ascent/Express Scripts/Cigna, and Zinc/CVS Caremark/Aetna, which collectively manage a significant portion of formulary decisions and reimbursement policies. These entities have the ability to deny or restrict formulary access to our products through various techniques, including National Drug Code (“NDC”) blocks, exclusion lists, and unfavorable tier placement. Such actions could severely limit patient access to our therapies, reduce sales, and negatively impact our financial performance. Additionally, the consolidation of PBMs and insurers further strengthens their negotiating leverage, increasing the risk of unfavorable pricing and reimbursement terms for our products.
Moreover, PBMs have significant influence over prescription drug access through formulary decisions, reimbursement policies, and utilization management practices. In some cases, PBMs may prioritize lower-cost alternatives, including off-label or unapproved therapies, over our FDA-approved products. This could include steering patients toward opioid-based treatments for fibromyalgia or barbiturates for migraine or other medications that do not meet the same regulatory and safety standards as our products. Such actions may limit patient access to clinically appropriate therapies and negatively impact our sales.
Healthcare legislative or regulatory reform measures, including government restrictions on pricing and reimbursement, may have a negative impact on our business and results of operations.
In the United States and some foreign jurisdictions, there have been, and continue to be, several legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing authorization of product candidates, restrict or regulate post-approval activities, and affect our ability to profitably sell any of our commercialized products.
Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives. For example, in the United States, the Patient Protection and Affordable Care Act (“ACA”), as supplemented by the Inflation Reduction Act of 2022 (“IRA”) and presidential executive orders, substantially changed the way healthcare is financed by both the government and private insurers, and significantly affects the pharmaceutical industry. Many provisions of the ACA impact the biopharmaceutical industry, including that in order for a biopharmaceutical product to receive federal reimbursement under the Medicare Part B and Medicaid programs or to be sold directly to U.S. government agencies, the manufacturer must extend discounts to entities eligible to participate in the drug pricing program under the Public Health Services Act, or PHS.
Additionally, the IRA, which is subject to active modification efforts, includes policies that have a direct impact on drug prices and reduced drug spending by the federal government. This legislation contains substantial drug pricing reforms, including the establishment of a drug price negotiation program within the U.S. Department of Health and Human Services that requires manufacturers to charge a negotiated “maximum fair price” for certain selected drugs covered by Medicare or pay an excise tax for noncompliance, rebate payment requirements on manufacturers of certain drugs payable under Medicare Parts B and D to penalize price increases that outpace inflation, and required discounts on Part D drugs. Moreover, presidential executive orders and agency guidance may rapidly change reimbursement and pricing conditions.
Legislative, administrative, and private payor efforts to control drug costs span a range of proposals, including drug price negotiation, Medicare Part D redesign, drug price inflation rebates, international mechanisms, generic drug promotion and anticompetitive behavior, manufacturer reporting, and reforms that could impact therapies utilizing the accelerated approval pathway. We cannot predict the ultimate content, timing or effect of any changes to the ACA, the Inflation Reduction Act, or other federal and state healthcare policy reform efforts including those aimed at drug pricing. There is no assurance that federal or state health care reform will not adversely affect our future business and financial results, and we cannot predict how future federal or state legislative, judicial or administrative changes relating to healthcare policy will affect our business.
At the state level, legislatures are increasingly passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price and patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Other examples of proposed and recent changes include, but are not limited to, expanding post-approval requirements, changing the Orphan Drug Act, restricting sales and promotional activities for pharmaceutical products, enacting drug price transparency laws, and the creation of prescription drug affordability boards with authority to review and, in some cases, constrain, payment levels.
We cannot be sure whether additional legislative or administrative changes will be enacted, or whether government regulations, guidance or interpretations will be changed, or what the impact of such changes would be on the marketing approvals, sales, pricing, or reimbursement of our drug candidates or products, if any, may be. We expect that these and other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved drug. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize our drugs.
65
In addition, FDA regulations and guidance may be revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. Any new regulations or guidance, or revisions or reinterpretations of existing regulations or guidance, may impose additional costs or lengthen FDA review times for our product candidates. Moreover, ongoing policy focus on accelerated approval oversight and facilitating the introduction of generics and biosimilars could tighten post-marketing obligations and increase competitive pressure on reference products. We cannot determine how changes in regulations, statutes, policies, or interpretations when and if issued, enacted or adopted, may affect our business in the future. Such changes could, among other things, require:
| ● | additional clinical trials to be conducted prior to obtaining approval; |
| ● | changes to manufacturing methods; |
| ● | increased competition; |
| ● | recalls, replacements, or discontinuance of one or more of our products; and |
| ● | additional recordkeeping. |
Such changes would likely require substantial time and impose significant costs, or could reduce the potential commercial value of our product candidates. In addition, delays in receipt of or failure to receive regulatory clearances or approvals for any other products would harm our business, financial condition, and results of operations.
Even if we obtain regulatory approval to market our product candidates, our product candidates may not be accepted by the market.
Even if the FDA approves one or more of our product candidates, physicians and patients may not accept or use it. Even if physicians and patients would like to use our products, our products may not gain market acceptance among healthcare payors such as managed care formularies, insurance companies or government programs such as Medicare or Medicaid. Acceptance and use of our products will depend upon a number of factors including: perceptions by members of the health care community, including physicians, about the safety and effectiveness of our drug or device product; cost-effectiveness of our product relative to competing products; availability of reimbursement for our product from government or other healthcare payors; and effectiveness of marketing and distribution efforts by us and our licensees and distributors, if any.
The degree of market acceptance of any pharmaceutical product that we develop will depend on a number of factors, including:
| ● | cost-effectiveness; |
| ● | the safety and effectiveness of our product candidates, including any significant potential side effects as compared to alternative products or treatment methods; |
| ● | the timing of market entry as compared to competitive products; |
| ● | the rate of adoption of our products by doctors and nurses; |
| ● | product labeling or product insert required by the FDA for each of our products; |
| ● | reimbursement policies of government and third-party payors; |
| ● | effectiveness of our sales, marketing and distribution capabilities and the effectiveness of such capabilities of our collaborative partners, if any; and |
| ● | unfavorable publicity concerning our products or any similar products. |
Because we expect sales of our current product candidates, if approved, to generate product revenues, the failure of these products to find market acceptance would harm our business and could require us to seek additional financing.
Uncertainty in Government funding and in-kind support policies and programs may adversely affect our business.
Changes in federal funding policies, including the ongoing review of DoD contracts and NIH grants and in-kind support by the current administration, could impact our financial resources and the progress of research conducted with U.S.-based university collaborators. While we have secured certain DoD and NIH funding and NIH in-kind support through Project NextGen, there is no guarantee that such funding or in-kind support will not be rescinded or otherwise restricted, particularly as the Biomedical Advanced Research and Development Authority (“BARDA”) has begun withdrawing or terminating some Project NextGen-related awards. Additionally, DoD, NIH and BARDA funding for any of our future projects may be delayed, reduced, or denied altogether. Any such changes could adversely affect our research programs, financial condition, and operational plans. Further, certain research projects conducted in collaboration with U.S.-based university collaborators could be slowed or discontinued.
66
RISKS RELATED TO PRODUCT DEVELOPMENT, REGULATORY APPROVAL, MANUFACTURING AND COMMERCIALIZATION
Our product candidates are novel and still in development.
Our drug development activities may not lead to commercially viable drugs for any of several reasons. For example, we may fail to identify appropriate targets or compounds, our drug candidates may fail to be safe and effective in clinical studies, or we may have inadequate financial or other resources to pursue development efforts for our drug candidates. Our drug candidates will require significant additional development, clinical studies, regulatory clearances and additional investment by us or our collaborators before they can be commercialized.
Further, we and our product candidates are subject to extensive regulation by the FDA and comparable regulatory authorities in other countries governing, among other things, research, testing, clinical studies, manufacturing, labeling, promotion, selling, adverse event reporting and recordkeeping. We are not permitted to market any of our product candidates in the United States until we receive approval of an NDA for a product candidate from the FDA or the equivalent approval from a foreign regulatory authority. Obtaining FDA approval is a lengthy, expensive and uncertain process. The success of our business depends on the successful development, approval and commercialization of our product candidates. Any projected sales or future revenue predictions are predicated upon FDA approval and market acceptance. If projected sales do not materialize for any reason, it would have a material adverse effect on our business and our ability to continue operations.
To obtain revenues from sales of our product candidates, we must succeed, either alone or with third parties, in developing, obtaining regulatory approval for, manufacturing and marketing drugs with commercial potential. We may never succeed in these activities, and we may not generate sufficient revenues to continue our business operations or achieve profitability.
If preclinical and nonclinical testing or clinical studies for our product candidates are unsuccessful or delayed, we will be unable to meet our anticipated development and commercialization timelines.
We rely and expect to continue to rely on third parties, including contract research organizations, or CROs, and outside consultants, to conduct, supervise or monitor some or all aspects of preclinical and nonclinical testing and clinical studies involving our product candidates. We have less control over the timing and other aspects of these preclinical and nonclinical testing activities and clinical studies than if we performed the monitoring and supervision entirely on our own. Third parties may not perform their responsibilities for our preclinical and nonclinical testing and clinical studies on our anticipated schedule or, for clinical studies, consistent with a clinical study protocol. Delays in preclinical and nonclinical testing, and clinical studies could significantly increase our product development costs and delay product commercialization. In addition, many of the factors that may cause, or lead to, a delay in the clinical studies may also ultimately lead to denial of regulatory approval of a product candidate.
The commencement of clinical studies can be delayed for a variety of reasons, including delays in:
| ● | demonstrating sufficient safety and efficacy to obtain regulatory approval to commence a clinical study; |
| ● | reaching agreement on acceptable terms with prospective CROs and study sites; |
| ● | developing a stable formulation of a product candidate; |
| ● | manufacturing sufficient quantities of a product candidate; and |
| ● | obtaining institutional review board, or IRB, approval to conduct a clinical study at a prospective site. |
Once a clinical study has begun, it may be delayed, suspended or terminated by us or the FDA or other regulatory authorities due to a number of factors, including:
| ● | ongoing discussions with the FDA or other regulatory authorities regarding the scope or design of our clinical studies; |
| ● | failure to conduct clinical studies in accordance with regulatory requirements; |
| ● | lower than anticipated recruitment or retention rate of patients in clinical studies; |
| ● | inspection of the clinical study operations or study sites by the FDA or other regulatory authorities resulting in the imposition of a clinical hold; |
| ● | lack of adequate funding to continue clinical studies; |
| ● | negative results of clinical studies; |
| ● | investigational drug product out-of-specification; or |
| ● | nonclinical or clinical safety observations, including adverse events and SAEs. |
67
If clinical studies are unsuccessful, and we are not able to obtain regulatory approvals for our product candidates under development, we will not be able to commercialize these products, and therefore may not be able to generate sufficient revenues to support our business.
We are subject to extensive and costly government regulation.
Product candidates employing our technology are subject to extensive and rigorous domestic government regulation including regulation by the FDA, the Centers for Medicare and Medicaid Services, other divisions of the United States Department of Health and Human Services, the United States Department of Justice, state and local governments, and their respective foreign equivalents. The FDA regulates the research, development, preclinical and nonclinical testing and clinical studies, manufacture, safety, effectiveness, record-keeping, reporting, labeling, storage, approval, advertising, promotion, sale, distribution, import, and export of biopharmaceutical products. The FDA regulates small molecule chemical entities as drugs, subject to an NDA under the FDCA. The FDA applies the same standards for biologics, requiring an IND application, followed by a Biologic License Application, or BLA, prior to licensure. Other products, such as vaccines, are also regulated under the Public Health Service Act. FDA has conflated the standards for approval of NDAs and BLAs so that they require the same types of information on safety, effectiveness, and CMCs. If products employing our technologies are marketed abroad, they will also be subject to extensive regulation by foreign governments, whether or not they have obtained FDA approval for a given product and its uses. Such foreign regulation may be equally or more demanding than corresponding United States regulation.
Government regulation substantially increases the cost and risk of researching, developing, manufacturing, and selling our products. The regulatory review and approval process, which includes preclinical and nonclinical testing and clinical studies of each product candidate, is lengthy, expensive, and uncertain. We or our collaborators must obtain and maintain regulatory authorization to conduct clinical studies. We or our collaborators must obtain regulatory approval for each product we intend to market, and the manufacturing facilities used for the products must be inspected and meet legal requirements. Securing regulatory approval requires the submission of extensive preclinical, nonclinical and clinical data and other supporting information for each proposed therapeutic indication in order to establish the product’s safety and efficacy, and in the case of biologics also potency and purity, for each intended use. The development and approval process takes many years, requires substantial resources, and may never lead to the approval of a product.
Even if we are able to obtain regulatory approval for a particular product, the approval may limit the indicated medical uses for the product, may otherwise limit our ability to promote, sell, and distribute the product, may require that we conduct costly post-marketing surveillance, and/or may require that we conduct ongoing post-marketing studies. Material changes to an approved product, such as, for example, manufacturing changes or revised labeling, may require further regulatory review and approval. Once obtained, any approvals may be withdrawn, including, for example, if there is a later discovery of previously unknown problems with the product, such as a previously unknown safety issue.
If we, our collaborators or our CMOs fail to comply with applicable regulatory requirements at any stage during the regulatory process, such noncompliance could result in, among other things, delays in the approval of applications or supplements to approved applications; refusal of a regulatory authority, including the FDA, to review pending market approval applications or supplements to approved applications; warning letters; fines; import and/or export restrictions; product recalls or seizures; injunctions; total or partial suspension of production; civil penalties; withdrawals of previously approved marketing applications or licenses; recommendations by the FDA or other regulatory authorities against governmental contracts; and/or criminal prosecutions.
Our product candidates may cause serious adverse events, or SAEs, or undesirable side effects which may delay or prevent marketing authorization, or, if approval is received, require them to be taken off the market, require them to include safety warnings or otherwise limit their sales.
SAEs or undesirable side effects from any of our other product candidates could arise either during clinical development or, if approved, after the approved product has been marketed. The results of future clinical studies may show that our product candidates cause SAEs or undesirable side effects, which could interrupt, delay or halt clinical studies, resulting in delay of, or failure to obtain, marketing authorization from the FDA and other regulatory authorities.
If any of our product candidates cause SAEs or undesirable side effects or suffer from quality control issues:
| ● | regulatory authorities may impose a clinical hold or risk evaluation and mitigation strategies, or REMS, which could result in substantial delays, significantly increase the cost of development, and/or adversely impact our ability to continue development of the product; |
68
| ● | regulatory authorities may require the addition of statements, specific warnings, or contraindications to the product label, or restrict the product’s indication to a smaller potential treatment population; |
| ● | we may be required to change the way the product is administered or conduct additional clinical studies; |
| ● | we may be required to implement a risk minimization action plan, which could result in substantial cost increases and have a negative impact on our ability to commercialize the product; |
| ● | we may be required to limit the participants who can receive the product; |
| ● | we may be subject to limitations on how we promote the product; |
| ● | we may, voluntarily or involuntarily, initiate field alerts for product recall, which may result in shortages; |
| ● | sales of the product may decrease significantly; |
| ● | regulatory authorities may require us to take our approved product off the market; |
| ● | we may be subject to litigation or product liability claims; and |
| ● | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the affected product or could substantially increase commercialization costs and expenses, which in turn could delay or prevent us from generating significant revenues from the sale of our products.
Any breakthrough, fast track or orphan drug designation or grant of priority review status by the FDA may not actually lead to a faster development or regulatory review or approval process, nor will it assure FDA approval of our product candidates. Additionally, our product candidates may treat indications that do not qualify for priority review vouchers.
If a product candidate offers major advances in treatment, the FDA may designate it eligible for priority review. The FDA has broad discretion whether or not to grant these designations, so even if we believe a particular product candidate is eligible for these designations, we cannot assure you that the FDA would decide to grant them. Even if we do receive fast track designation or priority review, we may not experience a faster development process, review or approval compared to conventional FDA procedures. The FDA may withdraw fast track designation if it believes that the designation is no longer supported by data from our clinical development program.
Even if approved, our product candidates will be subject to extensive post-approval regulation.
Once a product is approved, numerous post-approval requirements apply. Among other things, the holder of an approved NDA is subject to periodic and other FDA monitoring and reporting obligations, including obligations to monitor and report adverse events and instances of the failure of a product to meet the specifications in the NDA. Application holders must submit new or supplemental applications and obtain FDA approval for certain changes to the approved product, product labeling, or manufacturing process. Application holders must also submit advertising and other promotional material to the FDA and report on ongoing clinical studies.
Depending on the circumstances, failure to meet these post-approval requirements can result in criminal prosecution, fines, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of pre-marketing product approvals, or refusal to allow us to enter into supply contracts, including government contracts. In addition, even if we comply with FDA and other requirements, new information regarding the safety or effectiveness of a product could lead the FDA to modify or withdraw product approval.
Our relationships with customers, physicians and third-party payors is subject to federal and state healthcare fraud and abuse laws, false claims laws, health information privacy and security laws, and other healthcare laws and regulations. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
Healthcare providers, physicians and third-party payors in the United States and elsewhere will play a primary role in the recommendation and prescription of our products. Our current and future arrangements with healthcare professionals, principal investigators, consultants, customers and third-party payors subject us to various federal and state fraud and abuse laws and other health care laws, including, without limitation, the federal Anti-Kickback Statute, the federal civil and criminal false claims laws and the law commonly referred to as the Physician Payments Sunshine Act and regulations. These laws will impact, among other things, our clinical research, sales, marketing and educational programs. In addition, we may be subject to patient privacy laws by both the federal government and the states in which we conduct or may conduct our business. The laws that will affect our operations include, but are not limited to:
| ● | the federal Anti-Kickback Statute, which prohibits, among other things, persons or entities from knowingly and willfully soliciting, receiving, offering or paying any remuneration (including any kickback, bribe or rebate), directly or indirectly, overtly or covertly, in cash or in kind, in return for the purchase, recommendation, leasing or furnishing of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs; |
69
| ● | federal civil and criminal false claims laws, including, without limitation, the False Claims Act, and civil monetary penalty laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment or approval from Medicare, Medicaid or other government payors that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government; |
| ● | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created new federal criminal statutes that prohibit a person from knowingly and willfully executing a scheme or making false or fraudulent statements to defraud any healthcare benefit program, regardless of the payor (e.g., public or private); |
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and its implementing regulations, and as amended again by the final HIPAA omnibus rule, Modifications to the HIPAA Privacy, Security, Enforcement, and Breach Notification Rules Under HITECH and the Genetic Information Nondiscrimination Act; Other Modifications to HIPAA, published in January 2013, which imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information without appropriate authorization by entities subject to the rule, such as health plans, health care clearinghouses and health care providers, and their respective business associates; additionally, several states have enacted comprehensive privacy and data protection laws that may apply to health related and consumer data, some of which may not be protected health information under HIPAA, and other jurisdictions, including, among others the EU and UK, include strict rules on processing personal data, cross border data transfers, and data subject rights, and may impose significant fines and corrective orders; |
| ● | federal transparency laws, including the federal Physician Payments Sunshine Act, which is part of PPACA, that require certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to report annually to the Centers for Medicare & Medicaid Services, or CMS, information related to: (i) payments or other “transfers of value’’ made to covered recipients, including physicians, certain non physician practitioners, and teaching hospitals; and (ii) ownership and investment interests held by covered recipients and their immediate family members; |
| ● | state and foreign law equivalents of each of the above federal laws, state laws that require manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures, and state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or to adopt compliance programs as prescribed by state laws and regulations, or that otherwise restrict payments that may be made to healthcare providers; and |
| ● | state and foreign laws that govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws.
It is possible that government authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, disgorgement, imprisonment, exclusion of drugs from government funded healthcare programs, such as Medicare and Medicaid, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws and the curtailment or restructuring of our operations.
The risk of our being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, their provisions are open to a variety of interpretations, and they are subject to frequent policy changes. In addition, time-sensitive reporting and complex data aggregation requirements, and the need for ongoing training and monitoring create a shifting compliance environment. Efforts to ensure that our business complies with applicable healthcare laws and regulations involves substantial costs. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. The shifting compliance environment and the need to build and maintain robust and expandable systems to comply with multiple jurisdictions with different compliance and/or reporting requirements increases the possibility that we may run afoul of one or more of the requirements.
70
If we obtain approval to commercialize any approved products outside of the United States, a variety of risks associated with international operations could materially adversely affect our business.
If TONMYA or any of our product candidates is approved for commercialization outside of the United States, we intend to enter into agreements with third parties to market them on a worldwide basis or in more limited geographical regions. We expect that we will be subject to additional risks related to entering into international business relationships, including:
| ● | different regulatory requirements for drug approvals; |
| ● | reduced protection for intellectual property rights, including trade secret and patent rights; |
| ● | unexpected changes in tariffs, trade barriers and regulatory requirements; |
| ● | economic weakness, including inflation, or political instability in particular foreign economies and markets; |
| ● | compliance with tax, employment, immigration and labor laws for employees living or traveling abroad; |
| ● | foreign taxes, including withholding of payroll taxes; |
| ● | foreign currency fluctuations, which could result in increased operating expenses and reduced revenues, and other obligations incident to doing business in another country; |
| ● | workforce uncertainty in countries where labor unrest is more common than in the United States; |
| ● | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; |
| ● | business interruptions resulting from geopolitical actions, including war and terrorism, or natural disasters including earthquakes, hurricanes, floods and fires; and |
| ● | difficulty in importing and exporting clinical study materials and study samples. |
Governments outside the United States tend to impose strict price controls, which may adversely affect our revenues, if any.
If any our products are approved in foreign jurisdictions, we will be subject to pricing and reimbursement policies in those jurisdictions. In some countries, including countries in the EU, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a drug. To obtain reimbursement or pricing approval in some countries, governmental authorities adopt a number of different methodologies for assessing drug costs and reimbursement levels. These include comparisons with currently available medicines for the same indication and/or cost effectiveness assessments as the basis for negotiation. If reimbursement of our drugs is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business could be materially harmed.
If the FDA approves generic products that compete with TONMYA, sales of TONMYA would be adversely affected.
Once an NDA or marketing authorization application outside the United States is approved, the product covered thereby becomes a “listed drug” that can, in turn, be cited by potential competitors in support of approval of an abbreviated new drug application in the United States or equivalent marketing authorization application outside the United States. Agency regulations and other applicable regulations and policies provide incentives to manufacturers to create modified, non-infringing versions of a drug to facilitate the approval of an abbreviated new drug application or other application for generic substitutes in the United States and in nearly every pharmaceutical market around the world. These manufacturers might only be required to conduct a relatively inexpensive study to show that their product has the same active ingredient(s), dosage form, strength, route of administration and conditions of use, or labeling, as our product and that the generic product is bioequivalent to our product, meaning it is absorbed in the body at the same rate and to the same extent as our product. These generic equivalents, which must meet the same quality standards as branded pharmaceuticals, would be significantly less costly than our product to bring to market, and companies that produce generic equivalents are generally able to offer their products at lower prices. Thus, after the introduction of a generic competitor, a significant percentage of the sales of any branded product are typically lost to the generic product. Accordingly, competition from generic equivalents to TONMYA would materially adversely affect our future revenue, profitability and cash flows and substantially limit our ability to obtain a return on the investments we have made.
The biopharmaceutical industry is subject to extensive regulatory obligations and policies that may be subject to change, including due to judicial challenges.
The U. S. biopharmaceutical industry is highly regulated and subject to frequent and substantial changes, including as a result of new judicial or government actions. Legislative and regulatory agendas as they relate to the biopharmaceutical industry are currently uncertain. Changes in the regulatory approval process, or substantial reductions in the personnel who oversee that process, could affect our ability to obtain regulatory approval for our product candidates or the timeline in which we can obtain that approval. We and our current and future third-party collaborators rely on government programs and agencies, such as DTRA and the NIH, as a source of grant funding for scientific research relevant to our product candidates. Funding from government agencies such as DTRA and the NIH can fluctuate and is subject to the political process, which is often unpredictable, and the NIH and other research-funding agencies are revising how they make grant decisions, which may result in fewer grants. Reductions in government grants to us or our third-party collaborators may adversely impact our ability to develop our existing product candidates and our ability to identify new product candidates. In addition, on June 28, 2024, the U.S. Supreme Court issued an opinion holding that courts reviewing agency action pursuant to the Administrative Procedure Act (“APA”) “must exercise their independent judgment” and “may not defer to an agency interpretation of the law simply because a statute is ambiguous.” The decision could have a significant impact on how lower courts evaluate challenges to agency interpretations of law, including those by the FDA and other agencies with significant oversight and impact of the biopharmaceutical industry, and introduces additional uncertainty into the regulatory landscape for biopharmaceutical products. The new framework may increase both the frequency of such challenges and their odds of success by eliminating one way in which the government previously prevailed in such cases. As a result, significant regulatory policies could be subject to increased litigation and judicial scrutiny. We cannot predict how other future federal or state legislative or administrative changes relating to healthcare reform or the biopharmaceutical industry, or the regulatory agencies that oversee the biopharmaceutical industry, will affect our business.
71
Reductions in staffing and funding at FDA and other federal agencies could cause delays in the development and approval of our products.
Under the Federal Food, Drug, and Cosmetic Act, our products cannot be investigated in humans or marketed without approval from FDA. In addition, companies developing new therapies routinely seek and receive guidance from FDA regarding their methods and plans for developing their products. We and companies like us may also benefit from FDA-administered programs like orphan drug designation and expedited development pathways, e.g., breakthrough designation. Any material reductions in the ability of FDA to perform these and other functions may delay the development and approval of our product candidates.
Recent actions by the United States federal government have caused concern in the industry that this may occur. For example, beginning in February 2025, the Department of Health and Human Services initiated the termination of a large number of its probationary employees, a category that includes new federal employees and employees recently promoted or transferred to new positions or agencies. Larger layoffs may follow, according to a memorandum issued by the Office of Personnel Management in February 2025, and subsequent actions have, in fact, led to material workforce reductions at FDA. These terminations may significantly delay and impede our interactions with FDA, resulting in fewer or delayed meetings, slower feedback on development plans, less guidance issuance, greater variability in review times and possible delays in inspections or facility clearances. There are also reports that the United States federal government intends to request Congress to reduce FDA funding in upcoming budgets. Such funding cuts may also delay the development and approval of our products. Any significant decrease in FDA appropriations, or material reallocation of its budget or personnel, could impair the FDA’s ability to carry out its existing responsibilities and further slow the development, review, and potential approval of our product candidates. We cannot predict the scope, timing, outcome, or combined effect of these or any future federal actions affecting the FDA, but any significant adverse impact on the agency’s operations could materially and adversely affect our business and development timelines.
We may never receive regulatory approval to market our current or future product candidates outside of the U.S.
We plan to seek regulatory approval of our current or future product candidates outside of the U.S. In order to market any product outside of the U.S., however, we must establish and comply with the numerous and varying safety, efficacy and other regulatory requirements of other countries. Approval procedures vary among countries and can involve additional product candidate testing and additional administrative review periods. The time required to obtain approvals in other countries might differ substantially from that required to obtain FDA approval. The marketing approval processes in other countries generally implicate all of the risks detailed above regarding FDA approval in the U.S. as well as other risks. In particular, in many countries outside of the U.S., products must receive pricing and reimbursement approval before the product can be commercialized. Obtaining this approval can result in substantial delays in bringing products to market in such countries. Marketing approval in one country does not ensure marketing approval in another, but a failure or delay in obtaining marketing approval in one country may have a negative effect on the regulatory process in others. Failure to obtain marketing approval in other countries or any delay or other setback in obtaining such approval would impair our ability to market our current or future product candidates in such foreign markets. Any such impairment would reduce the size of our potential market, which could have a material adverse impact on our business, results of operations and prospects.
Our future growth may depend, in part, on our ability to penetrate foreign markets, where we would be subject to additional regulatory burdens and other risks and uncertainties that could materially adversely affect our business.
We are not permitted to market or promote any of our current or future product candidates before we receive regulatory approval from the applicable regulatory authority in that foreign market, and we may never receive such regulatory approval for any of our current or future product candidates. To obtain separate regulatory approval in many other countries we must comply with numerous and varying regulatory requirements of such countries regarding safety and efficacy and governing, among other things, clinical trials and commercial sales, pricing and distribution of our current or future product candidates, and we cannot predict success in these jurisdictions. If we obtain approval of our current or future product candidates and ultimately commercialize our current or future product candidates in foreign markets, we would be subject to additional risks and uncertainties, including:
| • | differing regulatory requirements in foreign countries, such that obtaining regulatory approvals outside of the U.S. may take longer and be more costly than obtaining approval in the U.S.; | |
| • | our customers’ ability to obtain reimbursement for our current or future product candidates in foreign markets; | |
| • | the burden of complying with complex and changing foreign regulatory, tax, accounting and legal requirements; | |
| • | different medical practices and customs in foreign countries affecting acceptance in the marketplace; | |
| • | import or export licensing requirements; | |
| • | longer accounts receivable collection times; | |
| • | longer lead times for shipping; | |
| • | language barriers for technical training; | |
| • | reduced protection of intellectual property rights in some foreign countries; | |
| • | the existence of additional potentially relevant third-party intellectual property rights; | |
| • | economic weakness, including inflation, or political instability in particular foreign economies and markets; | |
| • | compliance with tax, employment, immigration and labor laws for employees living or traveling abroad; | |
| • | foreign taxes, including withholding of payroll taxes; | |
| • | foreign currency fluctuations, which could result in increased operating expenses and reduced revenue, and other obligations incident to doing business in another country; | |
| • | difficulties staffing and managing foreign operations; | |
| • | workforce uncertainty in countries where labor unrest is more common than in the U.S.; | |
| • | potential liability under the Foreign Corrupt Practices Act of 1977 or comparable foreign regulations; | |
| • | the interpretation of contractual provisions governed by foreign laws in the event of a contract dispute; | |
| • | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and | |
| • | business interruptions resulting from geo-political actions, including war and terrorism. |
Foreign sales of our current or future product candidates could also be adversely affected by the imposition of governmental controls, political and economic instability, trade restrictions and changes in tariffs (including tariffs that have been or may in the future be imposed by the United States or other countries).
There may not be market interest in TNX-801.
The government is the only market for most medical countermeasures. This is because unlike other drugs and vaccines, these products are not sold to doctors, hospitals, or pharmacies. The BioShield Special Reserve Fund, or SRF, has been the sole medical countermeasures market for the last decade. The SRF is now appropriated annually and has not kept pace with the need for purchasing products ready for stockpiling. As such, even if TNX-801 were to receive FDA licensure, the commercial success of TNX-801 remains uncertain.
72
If technology developed for the purposes of developing new medicines or vaccines can be applied to the creation or development of biological weapons, then our technology may be considered “dual use” technology and be subject to limitations on public disclosure or export.
Our research and development of synthetic poxviruses is dedicated not only to creating tools that better protect public health but also to safeguarding any information with broad, dual-use potential that could be inappropriately applied. “Dual use research” is research conducted for legitimate purposes that generates knowledge, information, technologies, and/or products that can be reasonably anticipated to provide knowledge, information, products, or technologies that could be directly misapplied to pose a significant threat to public health, agricultural crops, or national security. Because variola, the agent that causes smallpox, is a pox virus, the technology we created could be considered dual use and could be subject to export control, for example under the Wassenaar Arrangement. Further, if federal authorities determine that our research is subject to institutional oversight, we will need to implement a risk-management plan developed in collaboration with the institutional review entity. Failure to comply with the plan may result in suspension, limitation, or termination of federal funding or loss of future federal funding opportunities for any of our research.
Competition and technological change may make our product candidates and technologies less attractive or obsolete.
We compete with established pharmaceutical and biotechnology companies that are pursuing other forms of treatment for the same or similar indications we are pursuing and that have greater financial and other resources. Other companies may succeed in developing products earlier than us, obtaining FDA approval for products more rapidly, or developing products that are more effective than our product candidates. Research and development by others may render our technology or product candidates obsolete or noncompetitive, or result in treatments or cures superior to any therapy we develop. We face competition from companies that internally develop competing technology or acquire competing technology from universities and other research institutions. As these companies develop their technologies, they may develop competitive positions that may prevent, make futile, or limit our product commercialization efforts, which would result in a decrease in the revenue we would be able to derive from the sale of any products.
There can be no assurance that any of our product candidates will be accepted by the marketplace as readily as these or other competing treatments. Furthermore, if our competitors’ products are approved before ours, it could be more difficult for us to obtain approval from the FDA. Even if our products are successfully developed and approved for use by all governing regulatory bodies, there can be no assurance that physicians and patients will accept our product(s) as a treatment of choice.
Additionally, if a competitor receives FDA approval before we do for a drug that is similar to one of our product candidates, FDA approval for our product candidate may be precluded or delayed due to periods of non-patent exclusivity and/or the listing with the FDA by the competitor of patents covering its newly-approved drug product. Periods of non-patent exclusivity for new versions of existing drugs such as TONMYA, can extend up to three and one-half years.
Furthermore, the pharmaceutical research industry is diverse, complex, and rapidly changing. By its nature, the business risks associated therewith are numerous and significant. The effects of competition, intellectual property disputes, market acceptance, and FDA regulations preclude us from forecasting revenues or income with certainty or even confidence.
RISKS RELATED TO OUR INTELLECTUAL PROPERTY RIGHTS AND REGULATORY EXCLUSIVITY
If we fail to protect our intellectual property rights, our ability to pursue the development of our technologies and products would be negatively affected.
Our success will depend in part on our ability to obtain patents and maintain adequate protection of our technologies and products. If we do not adequately protect our intellectual property, competitors may be able to use our technologies to produce and market drugs using our technologies and patents in direct competition with us and erode our competitive advantage. Some foreign countries lack rules and methods for defending intellectual property rights and do not protect proprietary rights to the same extent as the United States. Many companies have had difficulty protecting their proprietary rights in these foreign countries. We may not be able to prevent misappropriation of our proprietary rights and intellectual property rights in these and other countries.
We have received, and are currently seeking, patent protection for numerous compounds and methods of treating diseases. However, the patent process is subject to numerous risks and uncertainties, and there can be no assurance that we will be successful in protecting our products by obtaining and defending patents related to them. These risks and uncertainties include the following: patents that may be issued or licensed may be challenged, invalidated, or circumvented, or otherwise may not provide us any competitive advantage; our competitors, many of which have substantially greater resources than we and many of which have made significant investments in competing technologies, may seek, or may already have obtained, patents that will limit, interfere with, or eliminate our ability to make, use, and sell our potential products either in the United States or in international markets; there may be significant pressure on the United States government and other international governmental bodies to limit the scope of patent protection both inside and outside the United States for treatments that prove successful as a matter of public policy regarding worldwide health concerns; and countries other than the United States may have less robust patent laws than those upheld by United States courts, allowing foreign competitors the ability to exploit these laws to create, develop, and market competing products using our technologies and patents.
73
Moreover, any patent issued to us may not provide us with meaningful protection, or others may challenge, circumvent or narrow our patents. Third parties may also independently develop products similar to our products, duplicate our unpatented products or design around any patents or propriety technologies on products we develop. Additionally, extensive time is required for development, testing and regulatory review of a potential product. While extensions of patent terms due to regulatory delays may be available, it is possible that, before any of our product candidates can be commercialized, any related patent, even with an extension, may expire or remain in force for only a short period following commercialization, thereby reducing any advantages to us of the patent.
In addition, the PTO and patent offices in other jurisdictions have often required that patent applications concerning pharmaceutical and/or biotechnology-related inventions be limited or narrowed substantially to cover only the innovations specifically exemplified in the patent application, thereby limiting the scope of protection against competitive challenges. Thus, even if we or our licensors are able to obtain patents, the patents may be substantially narrower than anticipated.
Our success depends on our patents and patent applications that may be licensed exclusively to us and other patents and patent applications to which we may obtain assignment or licenses. We may not be aware, however, of all patents, published applications or published literature that may affect our business either by blocking our ability to commercialize our product candidates, by preventing the patentability of our product candidates to us or our licensors, or by covering the same or similar technologies. These patents, patent applications, and published literature may limit the scope of our future patent claims or adversely affect our ability to market our product candidates.
In addition to patents, we rely on a combination of trade secrets, confidentiality, nondisclosure and other contractual provisions, and security measures to protect our confidential and proprietary information. These measures may not adequately protect our trade secrets or other proprietary information. If they do not adequately protect our rights, third parties could use our technology, and we could lose any competitive advantage we may have. In addition, others may independently develop similar proprietary information or techniques or otherwise gain access to our trade secrets, which could impair any competitive advantage we may have.
Patent protection and other intellectual property protection is crucial to the success of our business and prospects, and there is a substantial risk that such protections will prove inadequate.
We are dependent on license relationships with third parties for certain of our drug development programs.
Companies that license technologies to us that we use in our research and development programs may require us to achieve milestones or devote minimum amounts of resources to develop products using those technologies. They may also require us to make significant royalty and milestone payments, including a percentage of any sublicensing income, as well as payments to reimburse them for patent costs. The number and variety of our research and development programs require us to establish priorities and to allocate available resources among competing programs. From time to time, we may choose to slow down or cease our efforts on particular products. If in doing so we fail to fully perform our obligations under a license, the licensor can terminate the license or permit our competitors to use the technology. Termination of these licenses or reduction or elimination of our licensed rights may result in our having to negotiate new or reinstated licenses with less favorable terms. Moreover, we may lose our right to market and sell any products based on the licensed technology. The occurrence of such events could materially harm our business.
We may be involved in lawsuits to protect or enforce our patents, which could be expensive and time consuming.
The pharmaceutical industry has been characterized by extensive litigation regarding patents and other intellectual property rights, and companies have employed intellectual property litigation to gain a competitive advantage. We may become subject to infringement claims or litigation arising out of present and future patents and other proceedings of our competitors. The defense and prosecution of intellectual property suits are costly and time-consuming to pursue, and their outcome is uncertain. Litigation may be necessary to determine the enforceability, scope, and validity of the proprietary rights of others. An adverse determination in litigation to which we may become a party could subject us to significant liabilities, require us to obtain licenses from third parties, or restrict or prevent us from selling our products in certain markets. Although patent and intellectual property disputes might be settled through licensing or similar arrangements, the costs associated with such arrangements may be substantial and could include our paying large, fixed payments and ongoing royalties. Furthermore, the necessary licenses may not be available on satisfactory terms or at all.
Competitors may infringe our patents, and we may file infringement claims to counter infringement or unauthorized use. Third parties may assert that our patents are invalid and/or unenforceable in these proceedings. Such litigation can be expensive, particularly for a company of our size, and time-consuming. In addition, in an infringement proceeding, a court may decide that a patent of ours is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover its technology. An adverse determination of any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly.
Third parties may also assert that our patents are invalid in patent office administrative proceedings. These proceedings include oppositions in the European Patent Office and inter partes review and post-grant review proceedings in the PTO. The success rate of these administrative challenges to patent validity in the United States is higher than it is for validity challenges in litigation.
Interference or derivation proceedings brought before the PTO may be necessary to determine priority of invention with respect to innovations disclosed in our patents or patent applications. During these proceedings, it may be determined that we do not have priority of invention for one or more aspects in our patents or patent applications and could result in the invalidation in part or whole of a patent or could put a patent application at risk of not issuing. Even if successful, an interference or derivation proceeding may result in substantial costs and distraction to our management.
74
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation or interference or derivation proceedings, there is a risk that some of our confidential information could be compromised by disclosure. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If investors perceive these results to be negative, the price of our common stock could be adversely affected.
Except for the oppositions to European Patents 2,968,992 and 2,683,245 (the Opposition Division in each of those oppositions maintained our claims in unamended form; Opponent did not appeal those decisions) and the opposition to European Patent 2,501,234 (the Opposition Division upheld the patent in unamended form; on appeal the Technical Board reversed and held the patent invalid, no appeal may be had from that decision), there are no unresolved communications, allegations, complaints or threats of litigation related to the possibility that our patents are invalid or unenforceable. Any litigation or claims against us, whether or not merited, may result in substantial costs, place a significant strain on our financial resources, divert the attention of management and harm our reputation. An adverse decision in litigation or administrative proceedings could result in inadequate protection for our product candidates and/or reduce the value of any license agreements we have with third parties.
If we infringe the rights of third parties we could be prevented from selling products, forced to pay damages, and defend against litigation.
If our products, methods, processes and other technologies infringe the proprietary rights of other parties, we could incur substantial costs and we may have to: obtain licenses, which may not be available on commercially reasonable terms, if at all; abandon an infringing product candidate; redesign our products or processes to avoid infringement; stop using the subject matter claimed in the patents held by others; pay damages; and/or defend litigation or administrative proceedings which may be costly whether we win or lose, and which could result in a substantial diversion of our financial and management resources.
There are risks to our intellectual property based on our international business initiatives.
We may face risks to our technology and intellectual property as a result of our conducting strategic business discussions outside of the United States, and particularly in jurisdictions that do not have comparable levels of protection of corporate proprietary information and assets such as intellectual property, trademarks, trade secrets, know-how and customer information and records. While these risks are common to many companies, conducting business in certain foreign jurisdictions, housing technology, data and intellectual property abroad, or licensing technology to joint ventures with foreign partners may have more significant exposure. For example, we have shared intellectual properties with entities in China pursuant to confidentiality agreements in connection with discussions on potential strategic collaborations, which may expose us to material risks of theft of our proprietary information and other intellectual property, including technical data, manufacturing processes, data sets or other sensitive information. For example, our technology may be reverse engineered by the parties or other parties, which could result in our patents being infringed or our know-how or trade secrets stolen. The risk can be by direct intrusion wherein technology and intellectual property is stolen or compromised through cyber intrusions or physical theft through corporate espionage, including with the assistance of insiders, or via more indirect routes.
We may be unable to protect the confidentiality of our trade secrets, thus harming our business and competitive position.
We rely upon trade secrets, including unpatented know-how, technology and other proprietary information, to develop and maintain our competitive position, which we seek to protect, in part, by confidentiality agreements with our employees and our collaborators and consultants. We also have agreements with our employees that obligate them to assign their inventions to us. However, it is possible that technology relevant to our business will be independently developed by a person that is not a party to such an agreement. Furthermore, if the employees, consultants or collaborators that are parties to these agreements breach or violate the terms of these agreements, we may not have adequate remedies for any such breach or violation, and we could lose our trade secrets through such breaches or violations. Further, our trade secrets could be disclosed, misappropriated or otherwise become known or be independently discovered by our competitors. In addition, intellectual property laws in foreign countries may not protect our intellectual property to the same extent as the laws of the United States. If our trade secrets are disclosed or misappropriated, it would harm our ability to protect our rights and have a material adverse effect on our business.
Third parties may assert that our employees or consultants have wrongfully used or disclosed confidential information or misappropriated trade secrets.
We employ individuals who were previously employed at universities or other diagnostic or biopharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees and consultants do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of a former employer or other third parties. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
We may need to license intellectual property from third parties, and such licenses may not be available or may not be available on commercially reasonable terms.
A third party may hold intellectual property, including patent rights, that are important or necessary to the development of our drug candidates. It may be necessary for us to use the patented or proprietary technology of a third party to commercialize our own technology or drug candidates, in which case we would be required to obtain a license from such third party. A license to such intellectual property may not be available or may not be available on commercially reasonable terms, which could have a material adverse effect on our business and financial condition.
GENERAL COMPANY-RELATED RISKS
We rely on third parties to conduct, supervise and monitor our clinical studies, and if those third parties perform in an unsatisfactory manner, it may harm our business.
We rely on CROs and clinical study sites to ensure the proper and timely conduct of our clinical studies. While we have agreements governing their activities, we will have limited influence over their actual performance. We will control only certain aspects of our CROs’ activities. Nevertheless, we will be responsible for ensuring that our clinical studies are conducted in accordance with the applicable protocol, legal, regulatory and scientific standards and our reliance on the CROs does not relieve us of our regulatory responsibilities.
We and our CROs are required to comply with the FDA’s cGCP for conducting, recording and reporting the results of clinical studies to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of clinical study participants are protected. The FDA enforces these cGCPs through periodic inspections of study sponsors, principal investigators and clinical study sites. If we or our CROs fail to comply with applicable cGCPs, the clinical data generated in our clinical studies may be deemed unreliable and the FDA may require us to perform additional clinical studies before approving any marketing applications. Upon inspection, the FDA may determine that our clinical studies did not comply with cGCPs. Accordingly, if our CROs fail to comply with these regulations, we may be required to repeat such clinical studies, which would delay the regulatory approval process.
75
Our CROs are not our employees, and we are not able to control whether or not they devote sufficient time and resources to our clinical studies. These CROs may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical studies, or other drug development activities which could harm our competitive position.
If our CROs do not successfully carry out their contractual duties or obligations, fail to meet expected deadlines, or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements, or for any other reasons, our clinical studies may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for, or successfully commercialize our product candidates. As a result, our financial results and the commercial prospects for such product candidates would be harmed, our costs could increase, and our ability to generate revenues could be delayed.
We also rely on other third parties to store and distribute drug products for our clinical studies. Any performance failure on the part of our distributors could delay clinical development or marketing authorization of our product candidates or commercialization of our products, if approved, producing additional losses and depriving us of potential product revenue.
In addition, we currently rely on foreign CROs and CMOs, and will likely continue to rely on foreign CROs and CMOs in the future. Foreign CMOs may be subject to U.S. legislation, including the proposed BIOSECURE Act, sanctions, trade restrictions and other foreign regulatory requirements which could increase the cost or reduce the supply of material available to us, delay the procurement or supply of such material or have an adverse effect on our ability to secure significant commitments from governments to purchase our potential therapies.
We may use our financial and human resources to pursue a particular research program or product candidate and fail to capitalize on programs or product candidates that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and human resources, we are currently focusing on development of our lead product candidates. As a result, we may forego or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on existing and future product candidates for specific indications may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through strategic alliance, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate, or we may allocate internal resources to a product candidate in a therapeutic area in which it would have been more advantageous to enter into a partnering arrangement.
We will need to expand our operations and increase the size of our company, and we may experience difficulties in managing growth.
As we increase our commercialization efforts for TONMYA and advance our product candidates through preclinical and nonclinical testing and clinical studies, and develop new product candidates, and buildout of our research and development and manufacturing facilities, we will need to increase our commercial, administrative, product development, scientific, regulatory and compliance headcount to manage these programs. In addition, to meet our obligations as a public company, we will need to increase our general and administrative capabilities. Our management, personnel and systems currently in place may not be adequate to support this future growth. Our need to effectively manage our operations, growth and various projects requires that we:
| ● | successfully attract and recruit new employees with the expertise and experience we will require; |
| ● | maintain a marketing, distribution and sales infrastructure in addition to a post-marketing surveillance program; |
| ● | manage our clinical programs effectively, which we anticipate being conducted at numerous clinical sites; and |
| ● | continue to improve our operational, manufacturing, quality assurance, financial and management controls, reporting systems and procedures. |
If we are unable to successfully manage this growth and increased complexity of operations, our business may be adversely affected.
Our executive officers and other key personnel are critical to our business, and our future success depends on our ability to retain them.
Our success depends to a significant extent upon the continued services of Dr. Seth Lederman, our President and Chief Executive Officer and Dr. Gregory M. Sullivan, our Chief Medical Officer. Dr. Lederman has overseen Tonix Pharmaceuticals, Inc., a wholly-owned subsidiary, since inception and provides leadership for our growth and operations strategy as well as being an inventor on many of our patents. Dr. Sullivan has served as our Chief Medical Officer since 2014 and directed the fibromyalgia program’s Phase 2 AtEase study, Phase 3 RELIEF study, the Phase 3 RALLY study and the Phase 3 RESILIENT study. Loss of the services of Drs. Lederman or Sullivan would have a material adverse effect on our growth, revenues, and prospective business. The loss of any of our key personnel, or the inability to attract and retain qualified personnel, may significantly delay or prevent the achievement of our research, development or business objectives and could materially adversely affect our business, financial condition and results of operations.
76
Any employment agreement we enter into will not ensure the retention of the employee who is a party to the agreement. In addition, we have only limited ability to prevent former employees from competing with us.
Furthermore, our future success will also depend in part on the continued service of our key scientific and management personnel and our ability to identify, hire, and retain additional personnel. We experience intense competition for qualified personnel and may be unable to attract and retain the personnel necessary for the development of our business. Moreover, competition for personnel with the scientific and technical skills that we seek is extremely high and is likely to remain high. Because of this competition, our compensation costs may increase significantly.
If we are unable to hire additional qualified personnel, our ability to grow our business may be harmed.
Over time we will need to hire additional qualified personnel with expertise in sales and marketing, drug development, product registration, clinical, preclinical and nonclinical research, quality compliance, government regulation, formulation and manufacturing and financial matters. We compete for qualified individuals with numerous biopharmaceutical companies, universities and other research institutions. Competition for such individuals is intense, and we cannot be certain that our search for such personnel will be successful. Attracting and retaining qualified personnel will be critical to our success.
We rely on third parties to manufacture our marketed products and the compounds used in our studies, and we intend to rely on them in the future. If these third parties do not manufacture our products and product candidates in sufficient quantities and at an acceptable cost, clinical development and commercialization of our products and product candidates could be delayed, prevented or impaired.
We rely on CMOs to manufacture all of our product candidates in clinical studies and our commercial products. Completion of our clinical studies and commercialization of our products requires the manufacture of a sufficient supply of our products. We have contracted with outside sources to manufacture our development compounds and commercial products. If, for any reason, we become unable to rely on our current manufacturing sources, either for clinical studies or for commercial quantities, then we would need to identify and contract with additional or replacement third-party manufacturers.
We believe that there are a variety of manufacturers that we may be able to retain to produce these products. However, once we retain a manufacturing source, if our manufacturers do not perform in a satisfactory manner, we may not be able to develop or commercialize our products as planned. Certain specialized manufacturers are expected to provide us with modified and unmodified pharmaceutical compounds, including finished products, for use in our preclinical and nonclinical testing and clinical studies and final product. Some of these materials are available from only one supplier or vendor. Any interruption in or termination of service by such sole source suppliers could result in a delay or interruption in manufacturing until we locate an alternative source of supply. Any delay or interruption in manufacturing operations (or failure to locate a suitable replacement for such suppliers) could materially adversely affect our business, prospects, or results of operations. We do not have any short-term or long-term manufacturing agreements with many of these manufacturers. If we fail to contract for manufacturing on acceptable terms or if third-party manufacturers do not perform as we expect, our development programs could be materially adversely affected, and our efforts to commercialize our marketed products will be materially impaired. This may result in delays in filing for and receiving FDA approval for one or more of our products and impair our revenues from sales. Any such delays could cause our prospects and financial condition to suffer significantly.
Failure by our third-party manufacturers to comply with the regulatory guidelines set forth by the FDA with respect to our products and product candidates could delay or prevent the completion of clinical studies, the approval of any product candidates or the commercialization of our products.
Third-party manufacturers must be inspected by FDA for cGMP compliance before they can produce commercial product. We may be in competition with other companies for access to these manufacturers’ facilities and may be subject to delays in manufacturing if the manufacturers give other clients higher priority than they give to us. If we are unable to secure and maintain third-party manufacturing capacity, the development and sales of our products and our financial performance may be materially affected.
Manufacturers are obligated to operate in accordance with FDA-mandated requirements. A failure of any of our third-party manufacturers to establish and follow cGMP requirements and to document their adherence to such practices may lead to significant delays in the availability of material for clinical studies, may delay or prevent filing or approval of marketing applications for our products, and may cause delays or interruptions in the availability of our products for commercial distribution following FDA approval. This could result in higher costs to us or deprive us of potential product revenues.
77
Drug manufacturers are subject to ongoing periodic unannounced inspections by the FDA, the Drug Enforcement Administration, or DEA, and corresponding state and foreign agencies to ensure strict compliance with cGMP requirements and other requirements under Federal drug laws, other government regulations and corresponding foreign standards. If we or our third-party manufacturers fail to comply with applicable regulations, sanctions could be imposed on us, including fines, injunctions, civil penalties, failure by the government to grant marketing approval of drugs, delays, suspension or withdrawal of approvals, seizures or recalls of product, operating restrictions and criminal prosecutions.
We are exposed to cybersecurity and data privacy risks that, if realized, could expose us to legal liability, damage our reputation and harm our business.
We face risks of cyber-attacks, computer hacks, theft, viruses, malicious software, phishing, employee error, denial-of-service attacks and other security breaches that could jeopardize the performance of our software and expose us to financial and reputational harm. Any of these occurrences could create liability for us, put our reputation in jeopardy and harm our business. Such harm could be in the form of theft of our or our customers’ confidential information, the inability to access our systems. In some cases, we rely on the safeguards put in place by third parties to protect against security threats. These third parties, including vendors that provide products and services for our operations, could also be a source of security risk to us in the event of a failure or a security incident affecting their own security systems and infrastructure. Our network of partners could also be a source of vulnerability to the extent their applications interface with ours, whether unintentionally or through a malicious backdoor. We do not review the software code included in third-party integrations in all instances. Because the techniques used to obtain unauthorized access or to sabotage systems change frequently and generally are not recognized until launched against a target, we or these third parties may be unable to anticipate these techniques or to implement adequate preventative measures. We have internal controls designed to prevent cyber-related frauds related to authorizing the transfer of funds, but such internal controls may not be adequate. With the increasing frequency of cyber-related frauds to obtain inappropriate payments and other threats related to cyber-attacks, we may find it necessary to expend resources to remediate cyber-related incidents or to enhance and strengthen our cybersecurity. Our remediation efforts may not be successful and could result in interruptions, delays or cessation of service. Although we have insurance coverage for losses associated with cyber-attacks, as with all insurance policies, there are coverage exclusions and limitations, and our coverage may not be sufficient to cover all possible claims, and we may still suffer losses that could have a material adverse effect on our reputation and business.
The increase in remote working arrangements by our employees, vendors, and other third parties also increase the risk of a data security compromise and the possible attack surfaces. Although we conduct training as part of our information security, cybersecurity, and data privacy efforts, that training cannot be completely effective in preventing those attacks from being successful. There can be no assurance that our cybersecurity risk management program and processes, including our policies, controls, or procedures, will be fully implemented, complied with or effective in protecting our systems and information.
Adverse global conditions, including economic uncertainty, may negatively impact our financial results.
Global conditions, dislocations in the financial markets, or inflation could adversely impact our business. In addition, the global macroeconomic environment has been and may continue to be negatively affected by, among other things, instability in global economic markets, increased U.S. trade tariffs and trade disputes with other countries, instability in the global credit markets, supply chain weaknesses, instability in the geopolitical environment, political tensions, and foreign governmental debt concerns. Such challenges have caused, and may continue to cause, uncertainty and instability in local economies and in global financial markets, which may adversely affect our business.
Our internal computer systems, or those of our CROs or other contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of our product development programs.
Despite the implementation of security measures, our internal computer systems and those of our CROs and other contractors and consultants are vulnerable to damage or disruption from computer viruses, software bugs, unauthorized access, natural disasters, terrorism, war, and telecommunication, equipment and electrical failures.
While we have not, to our knowledge, experienced any significant system failure, accident or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our programs. For example, the loss of clinical trial data from completed or ongoing clinical trials for any of our product candidates could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. Moreover, our information security systems and those of our CROs are also subject to laws and regulations requiring that we take measures to protect the privacy and security of certain information gathered and used in our business. For example, HIPAA and its implementing regulations impose, among other requirements, certain regulatory and contractual requirements regarding the privacy and security of personal health information. In the European Union the General Data Protection Regulation, or GDPR, is even more restrictive with respect to all personal information, including information masked by a coding system. In addition to HIPAA and GDPR, numerous other federal and state laws, including, without limitation, state security breach notification laws, state health information privacy laws and federal and state consumer protection laws, govern the collection, use, disclosure and storage of personal information. To the extent that any disruption or security breach results in a loss of or damage to our data or applications, or inappropriate disclosure or theft of confidential or proprietary information, we could incur liability, the further development of our product candidates could be delayed, our competitive position could be compromised, or our business reputation could be harmed.
78
Corporate and academic collaborators may take actions to delay, prevent, or undermine the success of our products.
Our operating and financial strategy for the development, clinical testing, manufacture, and commercialization of drug candidates is heavily dependent on our entering into collaborations with corporations, academic institutions, licensors, licensees, and other parties. Our current strategy assumes that we will successfully establish these collaborations, or similar relationships; however, there can be no assurance that we will be successful establishing such collaborations. Some of our existing collaborations are, and future collaborations may be, terminable at the sole discretion of the collaborator. Replacement collaborators might not be available on attractive terms, or at all. The activities of any collaborator will not be within our control and may not be within our power to influence. There can be no assurance that any collaborator will perform its obligations to our satisfaction or at all, that we will derive any revenue or profits from such collaborations, or that any collaborator will not compete with us. If any collaboration is not pursued, we may require substantially greater capital to undertake development and marketing of our proposed products and may not be able to develop and market such products effectively, if at all. In addition, a lack of development and marketing collaborations may lead to significant delays in introducing proposed products into certain markets and/or reduced sales of proposed products in such markets.
Data provided by collaborators and others upon which we rely that has not been independently verified could turn out to be false, misleading, or incomplete.
We rely on third-party vendors, scientists, and collaborators to provide us with significant data and other information related to our projects, clinical studies, and our business. If such third parties provide inaccurate, misleading, or incomplete data, our business, prospects, and results of operations could be materially adversely affected.
Our product candidates may face competition sooner than expected.
We intend to seek data exclusivity or market exclusivity for our product candidates provided under the FDCA and similar laws in other countries. We believe that TNX-801 could qualify for 12 years of data exclusivity under the Biologics Price Competition and Innovation Act of 2009, or BPCIA. Under the BPCIA, an application for a biosimilar product or BLA cannot be submitted to the FDA until four years, or if approved by the FDA, until 12 years, after the original brand product identified as the reference product is approved under a BLA. The BPCIA provides an abbreviated pathway for the approval of biosimilar and interchangeable biological products. The new abbreviated regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based on its similarity to an existing brand product. If the BPCIA is repealed or amended to shorten this exclusivity period, potentially creating the opportunity for biosimilar competition sooner than anticipated after the expiration of our patent protection, or business may be harmed. Moreover, the extent to which a biosimilar, once approved, will be substituted for any reference product in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing.
Our product candidates that are not, or are not considered, biologics that would qualify for exclusivity under the BPCIA may be eligible for market exclusivity as drugs under the FDCA. The FDCA provides a five-year period of non-patent marketing exclusivity within the U.S. to the first applicant to gain approval of an NDA for a new chemical entity (“NCE”). A drug is an NCE if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or ANDA, or a 505(b)(2) NDA, submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, for new indications, dosages, or strengths of an existing drug. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent.
79
Even if our product candidates are considered to be reference products eligible for 12 years of exclusivity under the BPCIA or five years of exclusivity under the FDCA, another company could market competing products if the FDA approves a full BLA or full NDA for such product containing the sponsor’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of the products. Moreover, an amendment or repeal of the BPCIA could result in a shorter exclusivity period for our product candidates, which could have a material adverse effect on our business.
If we are unable to establish effective marketing, sales and distribution capabilities or enter into agreements with third parties to market, sell and distribute our products, we may be unable to generate significant product awareness and that lack of awareness may limit the product revenues that we generate.
We recently expanded our commercial infrastructure for the marketing, sale, and distribution of pharmaceutical products, which includes a field-based sales force conducted through a combination of internal commercial personnel and contracted sales representatives to promote our commercial stage products throughout the United States. This effort requires additional compliance with a range of federal and state laws.
While many of our officers and employees have experience commercializing drug products with prior companies, as an organization we have limited experience in the marketing, sale, and distribution of pharmaceutical products, and there are significant risks involved in building, managing and maintaining a commercial infrastructure. We have to compete with other pharmaceutical and biotechnology companies to recruit, hire, train, manage, and retain marketing and sales personnel. In the event we are unable to maintain our marketing and sales infrastructure, we may not be able to successfully commercialize any of our existing commercial stage products or future product candidates, which would limit our ability to generate revenue. Factors that may inhibit our efforts to commercialize any of our products on our own include:
| ● | our inability to recruit, train, manage, and retain adequate numbers of effective sales and marketing personnel; |
| ● | the inability of sales personnel to obtain access to physicians or appropriately persuade adequate numbers of physicians to prescribe any of our current or future product candidates; |
| ● | our inability to effectively oversee a geographically dispersed sales and marketing team; |
| ● | the application of federal and state drug distribution and supply chain requirements to our business; |
| ● | the costs associated with training sales and marketing personnel on legal and regulatory compliance matters and monitoring their actions; |
| ● | an inability to secure adequate or any coverage and reimbursement by government and private health plans or other payers; |
| ● | the clinical indications and labeled claims for which the product is approved; |
| ● | limitations or warnings, including distribution or use restrictions, contained in the product’s approved labeling; |
| ● | any distribution and use restrictions imposed by the FDA or to which we agree as part of a mandatory REMS or voluntary risk management plan; |
| ● | liability for sales or marketing personnel who fail to comply with the applicable legal and regulatory requirements; |
| ● | the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; and |
| ● | unforeseen costs and expenses associated with creating an independent sales and marketing organization or engaging a contract sales organization. |
If additional product candidates are approved, we may incur expenses prior to product launch in expanding our sales force and compliant marketing and sales infrastructure. If a commercial launch is delayed as a result of FDA requirements or other reasons, we may incur these expenses prior to being able to realize any revenue from sales of such product candidate(s). Furthermore, our sales force and marketing teams may not be successful in commercializing any of our current or future product candidates.
We face the risk of product liability claims and may not be able to obtain insurance.
Our business exposes us to the risk of product liability claims that are inherent in the sale and development of drugs. If the use of one or more of our or our collaborators’ drugs harms people, we may be subject to costly and damaging product liability claims brought against us by clinical study participants, consumers, health care providers, pharmaceutical companies or others selling our products. Our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of pharmaceutical products we develop, alone or with collaborators. While we currently carry clinical study insurance and product liability insurance, we cannot predict all of the possible harms or side effects that may result and, therefore, the amount of insurance coverage we hold now or in the future may not be adequate to cover all liabilities we might incur. We expanded our insurance coverage to include the sale of TONMYA and our migraine products. However, if we are unable to maintain insurance at an acceptable cost or otherwise protect against potential product liability claims, we will be exposed to significant liabilities, which may materially and adversely affect our business and financial position. If we are sued for any injury allegedly caused by our or our collaborators’ products, our liability could exceed our total assets and our ability to pay the liability. A product liability claim or series of claims brought against us would decrease our cash and could cause our stock price to fall.
80
We use hazardous chemicals in our business. Potential claims relating to improper handling, storage or disposal of these chemicals could affect us and be time-consuming and costly.
Our research and development processes and/or those of our third-party contractors may involve the controlled use of hazardous materials and chemicals. These hazardous chemicals are reagents and solvents typically found in a chemistry laboratory. Our operations also produce hazardous waste products. Federal, state and local laws and regulations govern the use, manufacture, storage, handling and disposal of hazardous materials. While we attempt to comply with all environmental laws and regulations, including those relating to the outsourcing of the disposal of all hazardous chemicals and waste products, we cannot eliminate the risk of contamination from or discharge of hazardous materials and any resultant injury. In the event of such an accident, we could be held liable for any resulting damages and any liability could materially adversely affect our business, financial condition and results of operations.
Compliance with environmental laws and regulations may be expensive. Current or future environmental regulations may impair our research, development or production efforts. We might have to pay civil damages in the event of an improper or unauthorized release of, or exposure of individuals to, hazardous materials. We are not insured against these environmental risks.
If we enter into collaborations with third parties, they might also work with hazardous materials in connection with our collaborations. We may agree to indemnify our collaborators in some circumstances against damages and other liabilities arising out of development activities or products produced in connection with these collaborations.
In addition, the federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous or radioactive materials and waste products may require us to incur substantial compliance costs that could materially adversely affect our business, financial condition and results of operations.
Our insurance policies are expensive and protect us only from some business risks, which will leave us exposed to significant uninsured liabilities.
We carry insurance for most categories of risk that our business may encounter, however, we may not have adequate levels of coverage. We currently maintain general liability, clinical study, property, workers’ compensation, products liability and directors’ and officers’ insurance, along with an umbrella policy, which collectively costs approximately $1,500,000 per annum. We cannot provide any assurances that we will be able to maintain existing insurance at current or adequate levels of coverage. Any significant uninsured liability may require us to pay substantial amounts, which would adversely affect our cash position and results of operations.
If we retain collaborative partners and our partners do not satisfy their obligations, we will be unable to develop our partnered product candidates.
In the event we enter into any collaborative agreements, we may not have day-to-day control over the activities of our collaborative partners with respect to any of these product candidates. Any collaborative partner may not fulfill its obligations under these agreements. If a collaborative partner fails to fulfill its obligations under an agreement with us, we may be unable to assume the development of the products covered by that agreement or enter into alternative arrangements with a third party. In addition, we may encounter delays in the commercialization of the product candidate that is the subject of the agreement. Accordingly, our ability to receive any revenue from the product candidates covered by these agreements will be dependent on the efforts of our collaborative partner. We could also become involved in disputes with a collaborative partner, which could lead to delays in or termination of our development and commercialization programs and time-consuming and expensive litigation or arbitration. In addition, any such dispute could diminish our collaborators’ commitment to us and reduce the resources they devote to developing and commercializing our products. Conflicts or disputes with our collaborators, and competition from them, could harm our relationships with our other collaborators, restrict our ability to enter future collaboration agreements and delay the research, development or commercialization of our product candidates. If any collaborative partner terminates or breaches its agreement, or otherwise fails to complete its obligations in a timely manner, our chances of successfully developing or commercializing these product candidates would be materially and adversely affected. We may not be able to enter into collaborative agreements with partners on terms favorable to us, or at all. Our inability to enter into collaborative arrangements with collaborative partners, or our failure to maintain such arrangements, would limit the number of product candidates that we could develop and ultimately decrease our sources of any future revenues.
We face risks in connection with existing and future collaborations with respect to the development, manufacture, and commercialization of our product candidates.
We face a number of risks in connection with our current collaborations. Our collaboration agreements are subject to termination under various circumstances. Our collaborators may change the focus of their development and commercialization efforts or may have insufficient resources to effectively assist in the development of our products. Any future collaboration agreements may have the effect of limiting the areas of research and development that we may pursue, either alone or in collaboration with third parties. Further, disagreements with collaborators, including disagreements over proprietary rights, contract interpretation, or the preferred course of development, might cause delays, might result in litigation or arbitration, or might result in termination of the research, development or commercialization of our products. Any such disagreements would divert management attention and resources and be time-consuming and costly.
81
We face risks in connection with the testing, production and storage of our vaccine product candidates.
Developing our TNX-801 vaccine candidate requires testing of challenges with rabbitpox and vaccinia mpox viruses under controlled experimental conditions. The testing of TNX-801 may carry risk of infection and harm to individuals.
In addition, TNX-801 is a live form of the horsepox. We have initiated vaccine-manufacturing activities to support further nonclinical testing of TNX-801. The production and storage of the synthesized horsepox virus stock may carry risk of infection and harm to individuals. Any such infection could expose us to product and general liability claims, and may carry risk of infection and harm to individuals.
Changes in tax laws could adversely affect our business and financial condition.
Tax legislation continues to evolve globally with new laws and regulations that create uncertainty in the global economy. For example, on July 4, 2025, legislation commonly referred to as the One Big Beautiful Bill Act (the “OBBBA”) was signed into law, which includes significant provisions, including tax cut extensions and modifications to the international tax framework. We assessed the impact the OBBBA had on our financial condition, results of operations, cash flows or effective tax rate, and will continue to evaluate the impact of the legislative changes as additional guidance becomes available. Uncertainty remains regarding timing and interpretation by the tax authorities in the affected jurisdictions. Future tax reform and legislative changes could have an adverse impact on our future effective tax rate, tax liabilities, and cash tax.
RISKS RELATED TO OUR STOCK
Sales of additional shares of our common stock could cause the price of our common stock to decline.
Sales of substantial amounts of our common stock in the public market, or the availability of such shares for sale, by us or others, including the issuance of common stock upon exercise of outstanding options and warrants, could adversely affect the price of our common stock. We and our directors and officers may sell shares into the market, which could adversely affect the market price of shares of our common stock.
We may not be able to maintain compliance with the Listing Rules of the NASDAQ Stock Exchange.
There can be no assurance that in the future we will be able to maintain compliance with the Nasdaq Listing Rules, including the minimum bid price requirement and other applicable corporate governance requirements. If we fail to maintain compliance with the minimum bid requirement or to meet the other applicable continued listing requirements for the NASDAQ Global Select Market in the future and NASDAQ determines to delist our common stock, the delisting could adversely affect the market price and liquidity of our common stock and reduce our ability to raise additional capital. In addition, if our common stock is delisted from NASDAQ and the trading price remains below $5.00 per share, trading in our common stock might also become subject to the requirements of certain rules promulgated under the Exchange Act, which require additional disclosure by broker-dealers in connection with any trade involving a stock defined as a “penny stock” (generally, any equity security not listed on a national securities exchange or quoted on NASDAQ that has a market price of less than $5.00 per share, subject to certain exceptions).
An active trading market for our common stock may not be sustained.
Although our common stock is listed on the NASDAQ Global Select Market, the market for our shares has demonstrated varying levels of trading activity. Furthermore, the current level of trading may not be sustained in the future. The lack of an active market for our common stock may impair investors’ ability to sell their shares at the time they wish to sell them or at a price that they consider reasonable, may reduce the fair market value of their shares and may impair our ability to raise capital to continue to fund operations by selling shares and may impair our ability to acquire additional intellectual property assets by using our shares as consideration.
The market price of our common stock has been extremely volatile in the past and may be volatile in the future due to numerous circumstances beyond our control.
The market price of our common stock has fluctuated, and may fluctuate, widely, due to many factors, some of which may be beyond our control. These factors include, without limitation:
| ● | “short squeezes”; |
| ● | “short sellers”; |
| ● | comments by securities analysts or other third parties, including blogs, articles, message boards and social and other media; |
| ● | large stockholders exiting their position in our common stock or an increase or decrease in the short interest in our common stock; |
| ● | actual or anticipated fluctuations in our financial and operating results; |
| ● | the timing and allocations of new product candidates; |
| ● | public perception of our product candidates and competitive products; |
82
| ● | changes in financial estimates or recommendations by securities analysts; |
| ● | changes in the reimbursement policies of third party insurance companies or government agencies; and |
| ● | overall general market fluctuations. |
Stock markets in general and our stock price in particular have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies and our company. Broad market fluctuations may adversely affect the trading price of our common stock. In particular, a proportion of our common stock has been and may continue to be traded by short sellers which may put pressure on the supply and demand for our common stock, further influencing volatility in its market price. Additionally, these and other external factors have caused and may continue to cause the market price and demand for our common stock to fluctuate, which may limit or prevent investors from readily selling their shares of common stock and may otherwise negatively affect the liquidity of our common stock.
A “short squeeze” due to a sudden increase in demand for shares of our common stock that largely could lead to extreme price volatility in shares of our common stock.
Investors may purchase shares of our common stock to hedge existing exposure or to speculate on the price of our common stock. Speculation on the price of our common stock may involve long and short exposures. To the extent aggregate short exposure exceeds the number of shares of our common stock available for purchase on the open market, investors with short exposure may have to pay a premium to repurchase shares of our common stock for delivery to lenders of our common stock. Those repurchases may in turn, dramatically increase the price of our common stock until additional shares of our common stock are available for trading or borrowing. This is often referred to as a “short squeeze.” A proportion of our common stock has been and may continue to be traded by short sellers which may increase the likelihood that our common stock will be the target of a short squeeze. A short squeeze could lead to volatile price movements in shares of our common stock that are unrelated or disproportionate to our operating performance or prospectus and, once investors purchase the shares of our common stock necessary to cover their short positions, the price of our common stock may rapidly decline. Investors that purchase shares of our common stock during a short squeeze may lose a significant portion of their investment.
We do not anticipate paying dividends on our common stock and, accordingly, shareholders must rely on stock appreciation for any return on their investment.
We have never declared or paid cash dividends on our common stock and do not expect to do so in the foreseeable future. The declaration of dividends is subject to the discretion of our board of directors and will depend on various factors, including our operating results, financial condition, future prospects and any other factors deemed relevant by our board of directors. You should not rely on an investment in our company if you require dividend income from your investment in our company. The success of your investment will likely depend entirely upon any future appreciation of the market price of our common stock, which is uncertain and unpredictable. There is no guarantee that our common stock will appreciate in value.
The rights of the holders of common stock may be impaired by the potential issuance of preferred stock.
Our articles of incorporation give our board of directors the right to create new series of preferred stock. As a result, the board of directors may, without stockholder approval, issue preferred stock with voting, dividend, conversion, liquidation or other rights which could adversely affect the voting power and equity interest of the holders of common stock. Preferred stock, which could be issued with the right to more than one vote per share, could be utilized as a method of discouraging, delaying or preventing a change of control. The possible impact on takeover attempts could adversely affect the price of our common stock. Although we have no present intention to issue any shares of preferred stock or to create a series of preferred stock, we may issue such shares in the future.
If we fail to comply with the rules under the Sarbanes-Oxley Act of 2002 related to accounting controls and procedures, or if we discover material weaknesses and deficiencies in our internal control and accounting procedures, our stock price could decline significantly and raising capital could be more difficult.
If we fail to comply with the rules under the Sarbanes-Oxley Act of 2002 related to disclosure controls and procedures, or, if we discover material weaknesses and other deficiencies in our internal control and accounting procedures, our stock price could decline significantly and raising capital could be more difficult. Section 404 of the Sarbanes-Oxley Act requires annual management assessments of the effectiveness of our internal control over financial reporting. If material weaknesses or significant deficiencies are discovered or if we otherwise fail to achieve and maintain the adequacy of our internal control, we may not be able to ensure that we can conclude on an ongoing basis that we have effective internal controls over financial reporting in accordance with Section 404 of the Sarbanes-Oxley Act. Moreover, effective internal controls are necessary for us to produce reliable financial reports and are important to helping prevent financial fraud. If we cannot provide reliable financial reports or prevent fraud, our business and operating results could be harmed, investors could lose confidence in our reported financial information, and the trading price of our common stock could drop significantly.
83
If securities or industry analysts do not publish research or reports about our business, or if they change their recommendations regarding our stock adversely, our stock price and trading volume could decline.
The trading market for our common stock will be influenced by the research and reports that industry or securities analysts publish about us or our business. Our research coverage by industry and financial analysts is currently limited. Even if our analyst coverage increases, if one or more of the analysts who cover us downgrade our stock, our stock price would likely decline. If one or more of these analysts cease coverage of our company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause our stock price or trading volume to decline.
Other companies may have difficulty acquiring us, even if doing so would benefit our stockholders, due to provisions under our corporate charter and bylaws, as well as Nevada law.
Provisions in our articles of incorporation, our bylaws, and under Nevada law could make it more difficult for other companies to acquire us, even if doing so would benefit our stockholders. Our articles of incorporation and bylaws contain the following provisions, among others, which may inhibit an acquisition of our company by a third party:
| ● | advance notification procedures for matters to be brought before stockholder meetings |
| ● | a limitation on who may call stockholder meetings |
| ● | a limitation on the removal of directors |
| ● | the ability of our board of directors to issue up to 5,000,000 shares of preferred stock without a stockholder vote. |
We are also subject to provisions of Nevada law that prohibit us from engaging in any business combination with any “interested stockholder,” meaning generally that a stockholder who beneficially owns 10 percent or more of our stock cannot acquire us for a period of time after the date this person became an interested stockholder, unless various conditions are met, such as approval of the transaction by our board of directors and stockholders.
Our bylaws designate the Eighth Judicial District Court of Clark County, Nevada as the sole and exclusive forum for certain types of actions and proceedings that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers, employees or agents.
Our bylaws require that, to the fullest extent permitted by law, and unless the Company consents in writing to the selection of an alternative forum, the Eighth Judicial District Court of Clark County, Nevada, will, to the fullest extent permitted by law, be the sole and exclusive forum for each of the following:
| ● | any derivative action or proceeding brought in the name or right of the Company or on its behalf, |
| ● | any action asserting a claim for breach of any fiduciary duty owed by any director, officer, employee or agent of the Company to the Company or the Company’s stockholders, |
| ● | any action arising or asserting a claim arising pursuant to any provision of NRS Chapters 78 or 92A or any provision of our articles of incorporation or bylaws, or |
| ● | any action asserting a claim governed by the internal affairs doctrine, including, without limitation, any action to interpret, apply, enforce or determine the validity of our articles of incorporation or bylaws. |
Because the applicability of the exclusive forum provision is limited to the extent permitted by law, we believe that the exclusive forum provision would not apply to suits brought to enforce any duty or liability created by the Securities Exchange Act of 1934, as amended (Exchange Act), or any other claim for which the federal courts have exclusive jurisdiction, and that federal courts have concurrent jurisdiction over all suits brought to enforce any duty or liability created by the Securities Act of 1933, as amended (Securities Act). We note that there is uncertainty as to whether a court would enforce the provision and that investors cannot waive compliance with the federal securities laws and the rules and regulations thereunder. Although we believe this provision benefits us by providing increased consistency in the application of Nevada law in the types of lawsuits to which it applies, the provision may have the effect of discouraging lawsuits against our directors and officers.
ITEM 1B – UNRESOLVED STAFF COMMENTS
There are no unresolved staff comments at December 31, 2025.
84
ITEM 1C – CYBERSECURITY
Cybersecurity Risk Management
We face several cybersecurity risks in connection with our business. Our business strategy, results of operations, and financial condition have not, to date, been affected by risks from cybersecurity threats. During the reporting period, we have not experienced any material cyber incidents, nor have we experienced a series of immaterial incidents, which would require disclosure.
In the ordinary course of our business, we use, store and process data including data of our employees, partners, collaborators, and vendors. To effectively prevent, detect, and respond to cybersecurity threats, we maintain a cyber risk management program, which is comprised of a wide array of policies, standards, architecture, and processes. The cyber risk management program falls under the responsibility of the head of our Information Technology (“IT”), who has cross-functional expertise in IT, computer science, cyber security, and more than 20 years of experience. The IT head leads a team of IT specialists with similar IT and cybersecurity backgrounds. Under the guidance of the IT head, we develop, maintain, and evidence the policies, standards, and processes in a manner consistent with applicable legal requirements. We also utilize a variety of cybersecurity software from reputable vendors in cybersecurity.
Cybersecurity Governance
ITEM 2 – PROPERTIES
We maintain our principal office at 200 Connell Drive, Suite 3, Berkeley Heights, New Jersey 07922. Our telephone number at that office is (862) 799-8599 and our fax number is (212) 923-5700. On July 24, 2025, we entered into a lease, whereby we agreed to lease new office space, commencing November 2025 and expiring January 2030. In connection therewith, we maintain a letter of credit, which has a remaining balance of $386,000 as of December 31, 2025, and such amount is deposited into the restricted cash account maintained at the bank that issued the letter of credit.
We own and operate a research and development facility in Frederick, Maryland used for process development activities.
We own an approximately 44-acre site in Hamilton, Montana, for the construction of a vaccine development and commercial scale manufacturing facility. As of December 31, 2025, the facility was not ready for its intended use.
We own a 45,000 square foot facility in North Dartmouth, Massachusetts that houses our Advanced Development (“ADC”), for accelerated development and manufacturing of vaccines. The facility was decommissioned in 2024 decommissioned and may be reactivated on the earlier of 2027 or in the case of a national or international emergency.
85
Future minimum lease payments are as follows (in thousands):
| Year Ending December 31, | |||||
| 2026 | $ | 142 | |||
| 2027 | 480 | ||||
| 2028 | 451 | ||||
| 2029 | 366 | ||||
| 2030 and thereafter | 31 | ||||
| 1,470 | |||||
| Included interest | (161 | ) | |||
| $ | 1,309 | ||||
We believe that our existing facilities are suitable and adequate to meet our current business requirements.
ITEM 3 – LEGAL PROCEEDINGS
From time to time, we may become involved in various lawsuits and legal proceedings which arise in the ordinary course of business. However, litigation is subject to inherent uncertainties, and an adverse result in these or other matters may arise from time to time that may harm our business. We are currently not aware of any such legal proceedings or claims that we believe will have, individually or in the aggregate, a material adverse effect on our business, financial condition, operating results or cash flows.
ITEM 4 – MINE SAFETY DISCLOSURES
Not applicable.
PART II
ITEM 5 – MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock is listed on The NASDAQ Global Select Market under the symbol “TNXP”.
Holders
On March 11, 2026, the closing sale price of our common stock, as reported by The NASDAQ Stock Market, was $13.98 per share. On March 11, 2026, there were approximately 421 holders of record of our common stock. Because many of our shares of common stock are held by brokers and other institutions on behalf of stockholders, we are unable to estimate the total number of stockholders represented by these record holders.
Dividend Policy
We have never paid any cash dividends on our capital stock and do not anticipate paying any cash dividends on our common stock in the foreseeable future. We intend to retain future earnings to fund ongoing operations and future capital requirements of our business. Any future determination to pay cash dividends will be at the discretion of the Board and will be dependent upon our financial condition, results of operations, capital requirements and such other factors as the Board deems relevant.
Recent Sales of Unregistered Securities
None.
86
Repurchases of Equity Securities by the Issuer and Affiliated Purchasers
In November 2025, the Board of Directors approved a share repurchase program pursuant to which we may repurchase up to $35.0 million in value of our outstanding common stock from time to time on the open market and in privately negotiated transactions subject to market conditions, share price and other factors. Under this program, the timing and amount of any shares repurchased will be determined based on the Company’s evaluation of market conditions and other factors, and the program may be discontinued or suspended at any time. Repurchases will be made in accordance with the rules and regulations promulgated by the SEC and certain other legal requirements to which the Company may be subject. Repurchases may be made, in part, under a Rule 10b5-1 plan, which allows stock repurchases when the Company might otherwise be precluded from doing so.
The following table presents information with respect to our repurchases of common stock during the quarter ended December 31, 2025.
| Period | Total Number of Shares Purchased | Average Price Paid per Share |
Total Number of Shares Purchased as Part of Publicly Announced Programs | Approximate Dollar Value of Shares that May Yet Be Purchased Under Publicly Announced Programs |
|||||||||||
| October 1 - 31 | – | $ | – | – | $ | 29,051,125 | |||||||||
| November 1 - 30 | 66,000 | $ | 15.12 | 66,000 | $ | 28,051,060 | |||||||||
| December 1 - 31 | 381,903 | $ | 17.80 | 381,903 | $ | 21,239,960 | |||||||||
| Total | 447,903 | $ | 17.40 | 447,903 | |||||||||||
ITEM 6 – [RESERVED]
87
ITEM 7 – MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
This Management’s Discussion and Analysis of Financial Condition and Results of Operations includes a number of forward-looking statements that reflect Management’s current views with respect to future events and financial performance. You can identify these statements by forward-looking words such as “may” “will,” “expect,” “anticipate,” “believe,” “estimate” and “continue,” or similar words. Those statements include statements regarding the intent, belief or current expectations of us and members of its management team as well as the assumptions on which such statements are based and should be read together with the “Risk Factors” section of this Annual Report on Form 10-K for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis. Our actual results could differ materially from those anticipated in these forward-looking statements as a result of various factors, including those discussed below and elsewhere in this Annual Report and in other reports we file with the Securities and Exchange Commission, particularly those under “Risk Factors”.
We are a fully-integrated biopharmaceutical company commercializing and developing innovative therapies for central nervous system (“CNS”) disorders, immunology, infectious diseases, and rare diseases. Our portfolio consists of both commercial and development-stage programs.
In August 2025, we received approval from the FDA for TONMYA™ (cyclobenzaprine HCl sublingual tablets) for the treatment of fibromyalgia. TONMYA, our first internally developed product to become FDA approved, was commercially launched by us in the United States on November 17, 2025. TONMYA is the first new medicine for fibromyalgia in more than 15 years and is a centrally acting, non-opioid analgesic designed for bedtime administration and long-term use. The approval and launch of TONMYA marked major milestones in our evolution. We hold worldwide commercialization rights to TONMYA. In addition to TONMYA, we market two FDA-approved prescription products for the treatment of acute migraine: Zembrace® SymTouch® (sumatriptan injection) and Tosymra® (sumatriptan nasal spray). Our commercial platform includes sales, marketing, market access, distribution, and patient support capabilities.
We maintain a diversified development pipeline generated through internal discovery, in-licensing, acquisitions, and collaborations with academic and non-profit institutions. The Company’s pipeline addresses conditions that span central nervous system (“CNS”), infectious disease, immunology, and rare disease, with multiple programs in clinical and preclinical development. The proprietary cyclobenzaprine HCl sublingual tablet formulation contained in TONMYA is referred to as “TNX-102 SL” outside of the fibromyalgia indication. We are exploring the utility of TNX-102 SL (sublingual cyclobenzaprine) in Phase 2 clinical trials for major depressive disorder and acute stress disorder. TNX-102 SL is being developed to treat acute stress reaction and acute stress disorder under an Investigator-Initiated investigational new drug application (“IND”) at the University of North Carolina in the ongoing OASIS study funded by the U.S. Department of Defense (“DoD”). A Phase 2 study of TNX-102 SL for major depressive disorder is expected to commence mid-2026 under a Tonix IND that has been cleared by FDA.
Our clinical stage infectious disease portfolio includes monoclonal antibody TNX-4800 (anti-OspA from Borrelia burgdorferi) for seasonal prevention of Lyme disease, for which initiation of a Phase 2 field study is planned for the first half of 2027 and a Phase 2 human challenge study is planned for 2028, pending FDA clearances.
Our clinical-stage immunology development portfolio consists of biologics to address organ transplant rejection and autoimmunity, including TNX-1500, which is a Phase 2- ready Fc-modified humanized monoclonal antibody targeting CD40-ligand (CD40L or CD154) being developed for the prevention of allograft rejection and for the treatment of autoimmune diseases.
Another CNS candidate in clinical development is TNX-1300 (double-mutant cocaine esterase), which is in Phase 2 for the treatment of cocaine intoxication. TNX-1300 has been granted Breakthrough Therapy designation by the FDA.
Our clinical-stage rare disease portfolio includes TNX-2900, intranasal oxytocin potentiated with magnesium, in development for Prader-Willi syndrome and expected to start a Phase 2 study in the first quarter of 2027.
Our pre-clinical, pre-IND infectious disease portfolio includes TNX-801 (horsepox, live virus vaccine), as vaccine for mpox and smallpox. We own a facility in Dartmouth, MA that was purpose-built to manufacture TNX-801 under Good Manufacturing Practices (GMP) to support clinical development and potential commercialization. The facility was decommissioned in 2024 and may be reactivated on the earlier of 2027 or in the case of a national or international emergency.
Our pre-IND infectious disease portfolio also includes TNX-4200, which is a small molecule broad-spectrum antiviral agent targeting CD45 for the prevention or treatment of high lethality infections to improve the medical readiness of military personnel in biological threat environments. The TNX-4200 program is supported by a $34 million contract over five years from the U.S. DoD’s Defense Threat Reduction Agency (DTRA). We own and operate a state-of-the art research facility in Frederick, Maryland that supports this research.
Our pre-IND pre-clinical immunology portfolio includes TNX-1700, which is a fusion protein of TFF2 and albumin is in preclinical development for the treatment of gastric and colorectal cancer in combination with PD-1 blockade in collaboration with Columbia University.
Our pre-clinical, pre-IND CNS portfolio also includes TNX-4900, a highly selective small-molecule Sigma-1 receptor (“S1R”) antagonist for neuropathic pain.
Our product development candidates are investigational new drugs or biologics and have not been approved for any indication.
88
Zembrace SymTouch and Tosymra are registered trademarks of Tonix Medicines. TONMYA is a registered trademark of Tonix Pharma Limited. All other marks are the property of their respective owners. We are led by a management team with significant industry experience in drug development.
We complement our management team with a network of scientific, clinical, and regulatory advisors that includes recognized experts in their respective fields.
Results of Operations
We anticipate that our results of operations will fluctuate for the foreseeable future due to several factors, such as the sale of our commercialized assets, progress of our research and development efforts and the timing and outcome of regulatory submissions. Due to these uncertainties, accurate predictions of future operations are difficult or impossible to make.
Fiscal Year Ended December 31, 2025 Compared to Fiscal Year Ended December 31, 2024
The following table sets forth our operating expenses for the fiscal years ended December 31, 2025 and 2024 (in thousands):
| Year ended December 31, | ||||||||
| 2025 | 2024 | |||||||
| REVENUE | ||||||||
| Product revenue, net | $ | 13,107 | $ | 10,094 | ||||
| COSTS AND EXPENSES: | ||||||||
| Cost of sales | $ | 6,640 | $ | 7,765 | ||||
| Research and development | 44,486 | 39,972 | ||||||
| Selling, general and administrative | 87,684 | 40,101 | ||||||
| Asset impairment charges | — | 58,957 | ||||||
| Total operating expenses | 138,810 | 146,795 | ||||||
| Operating loss | (125,703 | ) | (136,701 | ) | ||||
| Grant income | 3,012 | 2,594 | ||||||
| Gain on change in fair value of warrant liabilities | — | 6,150 | ||||||
| Loss on extinguishment of debt | (2,092) | — | ||||||
| Interest income | 4,146 | 22 | ||||||
| Interest expense | (89 | ) | (1,234 | ) | ||||
| Other expense, net | (3,295 | ) | (867 | ) | ||||
| Net loss | $ | (124,021 | ) | $ | (130,036 | ) | ||
Revenues. Revenue recognized for the year ended December 31, 2025 and 2024 was $13.1 and $10.1 million, respectively.
The Company’s net product revenues are summarized below:
Year ended December 31, |
||||||||
| 2025 | 2024 | |||||||
| Tonmya | $ | 1,421 | $ | — | ||||
| Zembrace Symtouch | 9,314 | 8,546 | ||||||
| Tosymra | 2,372 | 1,548 | ||||||
| Total product revenues | $ | 13,107 | $ | 10,094 | ||||
Cost of Sales. Cost of goods sold during the year ended December 31, 2025, was $6.6 million, including write-downs related to Tosymra and Zembrace finished goods inventory of approximately $0.7 million based on an assessment of inventory on hand and projected sales prior to the respective expiration dates. Cost of sales recognized for the year ended December 31, 2024, was $7.8 million, including write-downs related to Tosymra and Zembrace finished goods inventory of approximately $1.5 million based on an assessment of inventory on hand and projected sales prior to the respective expiration dates.
Research and Development Expenses. Research and development expenses for the fiscal year ended December 31, 2025, were $44.5 million, an increase of $4.5 million, or 11%, from $40.0 million for the fiscal year ended December 31, 2024. This increase is predominately due to increased manufacturing expenses of $6.8 million and non-clinical expenses of $2.9 million as a result of pipeline prioritization period over period, and in employee-related expenses of $0.6 million due to increased headcount, offset by a decrease in regulatory expenses of $1.7 million and office-related expenses of $1.8 million due to a reduction in expenditures, as well as a decrease in clinical expenses of $2.0 million as a result of fewer clinical trials.
89
In August 2022, we received a Cooperative Agreement grant from the National Institute on Drug Abuse (“NIDA”), part of the National Institutes of Health, to support the development of its TNX-1300 product candidate for the treatment of cocaine intoxication. During the years ended December 31, 2025 and 2024, we recorded $0.6 and $1.6 million, respectively in funding as a reduction of related research and development expenses.
The table below summarizes our direct research and development expenses for our product candidates and development platform for the years ended December 31, 2025, and 2024.
December 31, (in thousands) |
||||||||||||
| 2025 | 2024 | Change | ||||||||||
| Research and development expenses: | ||||||||||||
| Direct expenses – TNX - 102 SL | $ | 5,601 | $ | 4,616 | $ | 985 | ||||||
| Direct expenses – TNX - 1500 | 5,810 | 2,772 | 3,038 | |||||||||
| Direct expenses – TNX - 801 | 2,038 | 599 | 1,439 | |||||||||
| Direct expenses – TNX - 1900 | 1,043 | 1,427 | (384 | ) | ||||||||
| Direct expenses – TNX - 4200 | 1,082 | — | 1,082 | |||||||||
| Direct expenses – TNX - 4800 | 1,458 | — | 1,458 | |||||||||
| Direct expenses – Other programs | 2,719 | 2,612 | 107 | |||||||||
| Internal staffing, overhead and other | 24,735 | 27,946 | (3,211 | ) | ||||||||
| Total research and development | $ | 44,486 | $ | 39,972 | $ | 4,514 | ||||||
Our direct research and development expenses consist principally of external costs for clinical, nonclinical, and manufacturing, such as fees paid to contractors, consultants and CROs in connection with our development work. Included in “Internal Staffing, Overhead and Other” is overhead, supplies, research and development employee costs (including stock option expenses), travel, regulatory and legal.
Selling, General and Administrative Expenses. Selling, general and administrative expenses for the fiscal year ended December 31, 2025, were $87.7 million, an increase of $47.6 million, or 119%, from $40.1 million incurred in the fiscal year ended December 31, 2024. The increase is primarily due to increase in sales and marketing of $37.7 million, an increase in professional legal fees of $1.5 million, and an increase in employee related costs of $7.6 million. All increases are related to our marketed migraine products as well as the launch of TONMYA in November 2025.
Asset impairment charges. We recognized a non-cash impairment charge of $48.8 million related to property and equipment, a non-cash impairment of $1.0 million related to goodwill, and a non-cash impairment charge of $9.2 million related to intangible assets, which is reflected in asset impairment charges in the consolidated statements of operations for the year ended December 31, 2024. No impairment charges were incurred during 2025.
The impairment of the Tosymra and Zembrace inventory, intangibles and goodwill was driven by our delayed investment in the sales personnel required to drive growth in the business as we are focusing our cash resources to further our efforts to bring TNX-102 SL through the approval process and to market. However, we believe that the benefits and long-term value proposition of the 2023 acquisition of Tosymra and Zembrace remain, in that we now have the infrastructure to be ready to manufacture and sell TNX-102 SL under an expedited timeline.
Net Loss. As a result of the foregoing, the net loss for the year ended December 31, 2025, was $124.0 million, compared to a net loss of $130.0 million for the year ended December 31, 2024.
License Agreement
On June 26, 2025, we obtained an exclusive worldwide license from the University of Massachusetts (“UMass”) Chan Medical School for the development of TNX-4800 (formerly known as mAb 2217LS). As of December 31, 2025, other than an upfront fee of $1.3 million, no payments have been accrued or paid in relation to this agreement.
90
Asset Purchase Agreements
On June 23, 2023, we entered into an asset purchase agreement with Upsher Smith for the acquisition of certain assets related to Zembrace and Tosymra.
We have assumed certain obligations of Upsher Smith, including the payment of quarterly royalty payments on annual net sales from the Business in the U.S. as follows: for Tosymra, 4% for net sales of $0 to $30 million, 7% of net sales of $30 to $75 million; 9% for net sales of $75 to $100 million; 12% for net sales of $100 to $150 million; and 15% for net sales greater than $150 million. Royalty payments with respect to Tosymra are payable until the expiration or termination of the product’s Orange Book listed patent(s) with respect to the United States or, outside the United States, the expiration of the last valid claim covering the product in the relevant country of the territory. For Zembrace, royalty payments on annual net sales in the U.S. are 3% for net sales of $0 to $30 million, 6% of net sales of $30 to $75 million; 12% for net sales of $75 to $100 million; 16% for net sales of greater than $100 million. Such royalty payments were payable until July 19, 2025. Upon the entry of a generic version of the relevant product, the applicable royalty rates will be reduced by 90% percent for Zembrace, and by 66.7% percent for Tosymra.
In addition, we have assumed the obligation to pay an additional 3% royalty on net sales of Tosymra, plus an additional 3% if a patent containing certain claims related to Tosymra issues in the U.S., for 15 years from the first commercial sale of Tosymra in the applicable country or for as long as the manufacture, use or sale of Tosymra in such country is covered by a valid claim of a licensed patent, and up to $15 million per Tosymra product on the achievement of sales milestones.
Liquidity and Capital Resources
As of December 31, 2025, we had working capital of $198.0 million, comprised primarily of cash and cash equivalents of $207.6 million, accounts receivable, net of $6.3 million, inventory of $6.0 million and prepaid expenses and other of $9.0 million, offset by $8.1 million of accounts payable, $22.6 million of accrued expenses, and current lease liabilities of $0.1 million. A significant portion of the accounts payable and accrued expenses are due to work performed in relation to our clinical programs, accruals for gross to net deductions related to our commercial products and product launch of TONMYA.
The following table provides a summary of operating, investing, and financing cash flows for the years ended December 31, 2025, and 2024, respectively (in thousands):
| December 31, | ||||||||
| 2025 | 2024 | |||||||
| Net cash used in operating activities | $ | (99,844 | ) | $ | (60,925 | ) | ||
| Net cash used in investing activities | (4,528 | ) | (120 | ) | ||||
| Net cash provided by financing activities | 214,530 | 134,872 | ||||||
For the years ended December 31, 2025, and 2024, we used approximately $99.8 million and $60.9 million in operating activities, respectively, which represents cash outlays for research and development and general and administrative expenses in such periods. The increase in cash outlays principally resulted from an increase in selling, general and administrative expenses as a result of the product launch of TONMYA. Cash used by investing activities for the year ended December 31, 2025, was approximately $4.5 million related to the issuance of a note and purchase of property and equipment repayment. Cash used by investing activities for the year ended December 31, 2024, was approximately $0.1 million related to the purchase of property and equipment.
For the year ended December 31, 2025, net proceeds from financing activities were $214.5 million, predominately from the sale of our common stock and warrants, which was offset by repurchase of common stock and repayment of debt. For the year ended December 31, 2024, net proceeds from financing activities were $134.9 million, primarily related to the sale of common stock and warrants.
We believe that our cash resources at December 31, 2025 and the proceeds that we raised from equity offerings in the first quarter of 2026, will meet our operating and capital expenditure requirements into the first quarter of 2027.
We continue to face significant challenges and uncertainties and must successfully launch TONMYA and obtain additional funding through public and private financing and collaborative arrangements with strategic partners to increase the funds available to fund operations. However, we may not be able to raise capital on terms acceptable to us, or at all. Without the successful product launch of TONMYA and obtaining additional funds, we may be forced to delay, scale back or eliminate some or all of our research and development activities or other operations, and potentially delay product development in an effort to maintain sufficient funds to continue operations. If any of these events occurs, our ability to achieve development and commercialization goals will be adversely affected and we may be forced to cease operations. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
91
Future Liquidity Requirements
We expect to incur losses from operations for the near future. We expect to increase our operating costs to align the Company’s capital and human resources with its previously announced strategic prioritization of the commercial launch of TONMYA for the treatment of fibromyalgia.
Our future capital requirements will depend on a number of factors, including the successful product launch of TONMYA, the progress of our research and development of product candidates, the timing and outcome of regulatory approvals, the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims and other intellectual property rights, the status of competitive products, the availability of financing and our success in developing markets for our product candidates.
We will need to successfully launch TONMYA and obtain additional capital in order to fund future research and development activities and future capital expenditures. Future financing may include the issuance of equity or debt securities, obtaining credit facilities, or other financing mechanisms. Even if we are able to raise the funds required, it is possible that we could incur unexpected costs and expenses, fail to collect significant amounts owed to us, or experience unexpected cash requirements that would force us to seek alternative financing. Furthermore, if we issue additional equity or debt securities, shareholders may experience additional dilution or the new equity securities may have rights, preferences or privileges senior to those of existing holders of our common stock.
If the product launch of TONMYA is unsuccessful and additional financing is not available or is not available on acceptable terms, we may be required to delay, reduce the scope of or eliminate our research and development programs, reduce our commercialization efforts or obtain funds through arrangements with collaborative partners or others that may require us to relinquish rights to certain product candidates that we might otherwise seek to develop or commercialize independently.
If additional financing is not available or is not available on acceptable terms, we may be required to delay, reduce the scope of or eliminate our research and development programs, reduce our commercialization efforts or obtain funds through arrangements with collaborative partners or others that may require us to relinquish rights to certain product candidates that we might otherwise seek to develop or commercialize independently.
December 2025 Financing
On December 29, 2025, we entered into a securities purchase agreement with an institutional investor, pursuant to which we sold 615,025 shares of common stock and pre-funded warrants to purchase up to 615,025 shares of common stock. The offering price per share of common stock was $16.26, and the offering price per share of pre-funded warrant was $16.259.
The offering closed on December 30, 2025. We incurred offering expenses of approximately $1.5 million, including placement agent fees of approximately $1.2 million. We received net proceeds of approximately $18.5 million, after deducting placement agent fees and other offering expenses.
2025 Lincoln Park Transaction
On June 11, 2025, we entered into a purchase agreement (the “2025 Purchase Agreement”) and a registration rights agreement (the “2025 Registration Rights Agreement”) with Lincoln Park. Pursuant to the terms of the 2025 Purchase Agreement, Lincoln Park has agreed to purchase us up to $75,000,000 of our common stock (subject to certain limitations) from time to time during the term of the 2025 Purchase Agreement. Pursuant to the terms of the 2025 Registration Rights Agreement, we filed with the SEC a registration statement to register for resale under the Securities Act the shares that have been or may be issued to Lincoln Park under the 2025 Purchase Agreement.
Pursuant to the terms of the 2025 Purchase Agreement, at the time we signed the 2025 Purchase Agreement and the 2025 Registration Rights Agreement, we issued 48,708 shares of common stock to Lincoln Park as consideration for its commitment to purchase shares of our common stock under the 2025 Purchase Agreement. The commitment shares were valued at $1.8 million and recorded as an addition to equity for the issuance of the common stock and treated as other expense, net on the consolidated statement of operations under the 2025 Purchase Agreement. No shares were sold during the year ended December 31, 2025, under the 2025 Purchase Agreement.
92
We evaluated the 2025 Purchase Agreement under ASC 815-40 Derivatives and Hedging-Contracts on an Entity’s Own Equity as it represents the right to require Lincoln Park to purchase shares of common stock in the future, similar to a put option. We concluded that the 2025 Purchase Agreement represents a freestanding derivative instrument that does not qualify for equity classification and therefore requires fair value accounting. We analyzed the terms of the contract and concluded that the derivative instrument had insignificant value as of December 31, 2025.
2025 At-the-Market Offering
On June 11, 2025, we entered into a Sales Agreement (the “2025 Sales Agreement”), with A.G.P./Alliance Global Partners (“AGP”) pursuant to which we may issue and sell, from time to time, shares of common stock having an aggregate offering price of up to $400.0 million in sales. AGP is sales agent under the ATM and paid a 3% commission on each sale under the 2025 Sales Agreement. Our common stock is sold at prevailing market prices at the time of the sale, and, as a result, prices will vary. During the year ended December 31, 2025, we sold 4.1 million shares of common stock under the 2025 Sales Agreement, for net proceeds of approximately $104.2 million. Subsequent to December 31, 2025, we sold 0.6 million shares of common stock under the 2025 Sales Agreement, for net proceeds of approximately $8.6 million.
2024 At-the-Market Offering
On July 30, 2024, we entered into a Sales Agreement (the “2024 Sales Agreement”), with AGP pursuant to which we could sell, from time to time, shares of common stock having an aggregate offering price of up to $250.0 million in sales. AGP is sales agent under the ATM and paid a 3% commission on each sale under the 2024 Sales Agreement. Our common stock is sold at prevailing market prices at the time of the sale, and, as a result, prices will vary. During the year ended December 31, 2025, we sold approximately 4.5 million shares of common stock under the Sales Agreement for net proceeds of approximately $112.9 million. During the year ended December 31, 2024, we sold approximately 4.2 million shares of common stock under the Sales Agreement, as defined below, for net proceeds of approximately $128.4 million. We can no longer sell shares under the 2024 Sales Agreement as the Company has reached the aggregate $250 million in sales.
July 2024 Financing
On July 9, 2024, we entered into a securities purchase agreement with certain institutional and retail investors, pursuant to which we sold 33,936 shares of common stock and pre-funded warrants to purchase up to 37,032 shares of common stock. The offering price per share of common stock was $57.00, and the offering price per share of pre-funded warrant was $56.99.
The offering closed on July 10, 2024. We incurred offering expenses of approximately $0.5 million, including placement agent fees of approximately $0.3 million. We received net proceeds of approximately $3.5 million, after deducting placement agent fees and other offering expenses.
June 2024 Financings
On June 12, 2024, we entered into a securities purchase agreement with certain investors, pursuant to which we sold 11,995 shares of common stock and pre-funded warrants to purchase up to 25,682 shares of common stock. The offering price per share of common stock was $106.50, and the offering price per share of pre-funded warrant was $106.40.
The offering closed on June 13, 2024. We incurred offering expenses of approximately $0.6 million, including placement agent fees of approximately $0.3 million. We received net proceeds of approximately $3.4 million, after deducting placement agent fees and other offering expenses.
On June 27, 2024, we entered into a securities purchase agreement with certain institutional and retail investors, pursuant to which we sold 28,339 shares of common stock and pre-funded warrants to purchase up to 42,282 shares of common stock. The offering price per share of common stock was $57.00, and the offering price per share of pre-funded warrant was $56.99.
The offering closed on June 28, 2024. We incurred offering expenses of approximately $0.6 million, including placement agent fees of approximately $0.3 million. We received net proceeds of approximately $3.4 million, after deducting placement agent fees and other offering expenses.
93
March 2024 Financing
On March 28, 2024, we entered into an agreement to sell 3,365 shares of common stock, pre-funded warrants to purchase up to 1,219 shares of common stock, and accompanying Series E warrants to purchase up to 4,584 shares of common stock with an exercise price of $1,056.00 per share and expiring five and a half years from date of issuance in a public offering, which closed on April 1, 2024. The offering price per share of common stock was $960.00, and the offering price per share of pre-funded warrants was $959.68.
We incurred expenses of approximately $0.5 million, including placement agent fees of approximately $0.3 million. We received net proceeds of approximately $3.9 million, after deducting placement agent fees and other offering expenses.
Additionally, with the closing of the financing on April 1, 2024, we entered into warrant amendments (collectively, the “Warrant Amendments”) with certain holders of its common warrants (referred to herein as the “Existing Warrants”). We agreed to amend the exercise price of each Existing Warrant to $1,056.00 upon approval by our stockholders of a proposal to allow the Existing Warrants to become exercisable in accordance with Nasdaq Listing Rule 5635 or, if stockholder approval is not obtained by October 1, 2024, we agreed to automatically amend the exercise price of the Existing Warrants to the Minimum Price (as defined in Nasdaq Listing Rule 5635(d)) of our common stock on October 1, 2024, if and only if the Minimum Price is below the then current exercise price. Upon stockholder approval, the termination date for the warrants issued August 2023 (the “August Warrants”) to purchase up to an aggregate of 2,172 shares was amended to April 1, 2029; the termination date for Series A Warrants to purchase up to an aggregate of approximately 2,782 shares is April 1, 2029; the termination date for Series B Warrants to purchase up to an aggregate of approximately 2,782 shares is April 1, 2025; the termination date for Series C Warrants to purchase up to an aggregate of approximately 10,884 shares is the earlier of (i) April 1, 2026 and (ii) 10 trading days following notice by the Company to the Series C Warrant holders of our public announcement of the FDA’s acknowledgement and acceptance of the Company’s NDA relating to TNX-102 SL in patients with Fibromyalgia; the termination date for Series D Warrants to purchase up to an aggregate of approximately 10,884 shares is April 1, 2029. The other terms of the Existing Warrants remained unchanged.
We evaluated the Warrant Amendments as of April 1, 2024, and determined that the potential adjustment to the exercise price that is contingent on stockholder approval precluded the Existing Warrants from being indexed to our own stock, and as a result, did not meet the criteria for equity classification under ASC 815-40. We accounted for the incremental fair value of the Warrant Amendments of $3.0 million as a direct and incremental cost of the March 2024 financing as an offset to the proceeds received. As all of the Existing Warrants were equity-classified prior to the Warrant Amendments, the net impact to the consolidated statement of stockholders’ equity was zero. We then reclassified the Existing Warrants from equity to liabilities at post-modification fair value on April 1, 2024. On May 22, 2024, the date of our stockholders approved the proposal to fix the exercise prices at $1,056.00 per share, the Existing Warrants were adjusted to fair value and reclassified back to equity.
The liability-classified Series D Warrants and all of the Series C Warrants were presented within non-current liabilities on the consolidated balance sheets as of December 31, 2023, and were adjusted to fair value through January 25, 2024, when the warrants were reclassified to equity. Changes in the fair value of the liability-classified warrants were recognized as a separate component in the consolidated statement of operations.
Stock Repurchases
In September 2024, the Board of Directors approved a 2024 share repurchase program pursuant to which we may repurchase up to $10.0 million in value of its outstanding common stock from time to time on the open market and in privately negotiated transactions subject to market conditions, share price and other factors. In November 2025, the amount increased to $35.0 million.
During the year ended December 31, 2025, we repurchased 847,903 shares of its common stock outstanding under the 2024 share repurchase at prices ranging from $9.98 to $20.47 per share for a gross aggregate cost of approximately $13.8 million. The repurchased shares were immediately retired.
The timing and amount of any shares repurchased will be determined based on our evaluation of market conditions and other factors and the share repurchase program may be discontinued or suspended at any time. Repurchases will be made in accordance with the rules and regulations promulgated by the Securities and Exchange Commission and certain other legal requirements to which we may be subject. Repurchases may be made, in part, under a Rule 10b5-1 plan, which allows stock repurchases when we might otherwise be precluded from doing so.
Debt Financing
On December 8, 2023, we executed a Loan and Guaranty Agreement (the “Loan Agreement”) to issue a 36-month term loan (the “Term Loan”) in the principal amount of $11.0 million with a maturity date of December 8, 2026 (the “Maturity Date”). The Term Loan was funded with an original issue discount of 9% of the principal amount of the Term Loan, or $1.0 million, which was being amortized over the term of the debt as an adjustment to the effective interest rate on the outstanding borrowings.
94
Borrowings under the Term Loan bear interest at a fluctuating rate equal to the greater of (i) the prime rate as defined in the Loan Agreement plus 3.5% and (ii) 12%. Interest was payable monthly in arrears commencing in December 2023. In connection with the Term Loan, we deposited into a reserve account $1.8 million to be used exclusively to fund interest payments related to the Term Loan. The deposit is reflected as prepaid and other current assets on the consolidated balance sheet.
Commencing on March 8, 2024 and continuing monthly through the Maturity Date, the outstanding principal will be due and payable in monthly installments of $0.2 million, with the final remaining balance of unpaid principal and interest due and payable on the Maturity Date. In addition, we paid a monthly collateral monitoring charge equal to 0.23% of the outstanding principal amount of the term loan as of the date of payment. We incurred $1.1 million in issuance costs, which was amortized over the term of the debt as an adjustment to the effective interest rate on the outstanding borrowings.
The Loan Agreement provides for voluntary prepayments of the Term Loan, in whole or in part, subject to a prepayment premium. The Loan Agreement contains customary affirmative and negative covenants by us, which among other things, will require us to provide certain financial reports to the lenders, to maintain a deposit account to fund interest payments, and limit the ability of us to incur or guarantee additional indebtedness, pay dividends or make other equity distributions, sell assets, engage in certain transactions, and effect a consolidation or merger. Our obligations under the Loan Agreement may be accelerated upon customary events of default, including non-payment of principal, interest, fees and other amounts, covenant default, insolvency, material judgements, inaccuracy of representations and warranties, invalidity of guarantees. The Term Loan was secured by first priority security interests in our R&D Center in Frederick, Maryland, the Advanced Development Center in North Dartmouth, Massachusetts, and substantially all of the relevant deposit accounts.
During the first quarter of 2025, we paid $9.6 million as a result of a pay-off of the above-mentioned loan. The pay-off amount paid by us in connection with the termination of the Loan Agreement was pursuant to a pay-off letter and includes a prepayment fee of $1.0 million in accordance with the terms and provisions of the Loan Agreement.
Stock Compensation
On May 1, 2020, our stockholders approved the Tonix Pharmaceuticals Holding Corp. Amended and Restated 2020 Stock Incentive Plan, and May 8, 2025, our stockholders approved an amendment to this plan (as amended, “Amended and Restated 2020 Plan”).
Under the terms of the Amended and Restated 2020 Plan, we may issue (1) stock options (incentive and nonstatutory), (2) restricted stock, (3) stock appreciation rights (“SARs”), (4) RSUs, (5) other stock-based awards, and (6) cash-based awards. The Amended and Restated 2020 Plan initially provided for the issuance of up to 50,000 shares of common stock, which amount will be increased to the extent that awards granted under the Plans are forfeited, expire or are settled for cash (except as otherwise provided in the Amended and Restated 2020 Plan). In addition, the Amended and Restated 2020 Plan contains an “evergreen provision” providing for an annual increase in the number of shares of our common stock available for issuance under the Amended and Restated 2020 Plan on January 1 of each year for a period of ten years, commencing on January 1, 2021 and ending on (and including) January 1, 2030, in an amount equal to the difference between (x) twenty percent (20%) of the total number of shares of common stock outstanding on December 31st of the preceding calendar year, and (y) the total number of shares of common stock reserved under the Amended and Restated 2020 Plan on December 31st of such preceding calendar year (including shares subject to outstanding awards, issued pursuant to awards or available for future awards). On May 8, 2025, our stockholders approved the addition of 1,000,000 shares to the Company’s Amended and Restated 2020 Plan.
The Board of Directors determines the exercise price, vesting and expiration period of the grants under the Amended and Restated 2020 Plan. However, the exercise price of an incentive stock option may not be less than 110% of fair value of the common stock at the date of the grant for a 10% or more shareholder and 100% of fair value for a grantee who is not a 10% shareholder. The fair value of the common stock is determined based on quoted market price or in absence of such quoted market price, by the Board of Directors in good faith. Additionally, the expiration period of grants under the Amended and Restated 2020 Plan may not be more than ten years. As of December 31, 2025, there were 726,433 options available for future grants under the Amended and Restated 2020 Plan.
The aggregate intrinsic value in the preceding table represents the total pretax intrinsic value, based on options with an exercise price less than the Company’s closing stock price at the respective dates.
The weighted average fair value of options granted during the year ended December 31, 2025, and December 31, 2024 was $13.10 and $868.00 per share, respectively.
95
We measure the fair value of stock options on the date of grant, based on the Black Scholes option pricing model using certain assumptions discussed below, and the closing market price of our common stock on the date of the grant. The fair value of the award is measured on the grant date. One-third of most stock options granted pursuant to the Plans vest 12 months from the date of grant and 1/36th each month thereafter for 24 months and expire ten years from the date of grant. In addition, we issue options to directors which vest over a one-year period. We also issue premium options to executive officers which have an exercise price greater than the grant date fair value and has issued performance-based options which vest when target parameters are met or probable of being met, subject in each case to a one year minimum service period prior to vesting. Stock-based compensation expense related to awards is amortized over the applicable service period using the straight-line method.
The risk-free interest rate is based on the yield of Daily U.S. Treasury Yield Curve Rates with terms equal to the expected term of the options as of the grant date. The expected term of options is determined using the simplified method, as provided in an SEC Staff Accounting Bulletin, and the expected stock price volatility is based on our historical stock price volatility.
Stock-based compensation expense relating to options granted of $6.0 million, of which $4.1 million and $1.9 million, related to General and Administration and Research and Development, respectively was recognized for the year ended December 31, 2025. Stock-based compensation expense relating to options granted of $4.8 million, of which $3.4 million and $1.4 million, related to General and Administration and Research and Development, respectively was recognized for the year ended December 31, 2024.
As of December 31, 2025, we had approximately $12.3 million of total unrecognized compensation cost related to non-vested awards granted under the Plans, which we expect to recognize over a weighted average period of 2.82 years.
Commitments
Research and Development Contracts
We have entered into contracts with various contract research organizations with outstanding commitments aggregating approximately $52.3 million at December 31, 2025 for future work to be performed.
We have entered into various exclusive license agreements with various institutions with the right to sublicense, certain patents, technical information and material, and to develop and commercialize products thereunder. In addition to any upfront payments already paid, we may be obligated to pay milestone fees ranging from $25,000 to $5.0 million based on the potential achievement of certain development milestones, as well as milestone fees ranging from $55,000 to $20.0 million based on certain potential commercial achievements, as specified in the respective license agreement. Additionally, for licensed products sold during the applicable royalty term, we must pay royalties in the low-to-mid single digits, beginning in the year after we complete our first commercial sale of a licensed product. Finally, we have the right to grant sublicenses to third parties under each license agreement and is required to pay a sublicense income share based on the stage of development of the licensed product at the time the sublicense is granted.
Operating leases
As of December 31, 2025, future minimum lease payments are as follows (in thousands):
| Year Ending December 31, | |||||
| 2026 | $ | 142 | |||
| 2027 | 480 | ||||
| 2028 | 451 | ||||
| 2029 | 366 | ||||
| 2030 and thereafter | 31 | ||||
| 1,470 | |||||
| Included interest | (161 | ) | |||
| $ | 1,309 | ||||
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses. We evaluate our estimates and judgments on an ongoing basis. We base our estimates on historical experience and on assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates.
96
We believe the following critical accounting policies affect our more significant judgments and estimates used in the preparation of our consolidated financial statements.
Revenue Recognition. Our gross product revenues are subject to a variety of deductions, which generally are estimated and recorded in the same period that the revenues are recognized. Such variable consideration represents chargebacks, rebates, prompt pay and other sales discounts, and product returns. These deductions represent estimates of the related obligations and, as such, knowledge and judgment are required when estimating the impact of these revenue deductions on gross sales for a reporting period. We began recognizing revenue following the completion of the USL Acquisition, beginning July 1, 2023, and required variable consideration estimates are currently primarily based on the acquired products historical results. Adjustments to these estimates to reflect actual results or updated expectations will be assessed each period. If any of our ratios, factors, assessments, experiences, or judgments are not indicative or accurate estimates of our future experience, our results could be materially affected. The potential of our estimates to vary differs by program, product, type of customer and geographic location. In addition, estimates associated with U.S. Medicare and Medicaid governmental rebate programs are at risk for material adjustment because of the extensive time delay.
Research and Development. We outsource certain of our research and development efforts and expense the related costs as incurred, including the cost of manufacturing product for testing, licensing fees and costs associated with planning and conducting clinical trials. The value ascribed to patents and other intellectual property acquired was expensed as research and development costs, as it related to particular research and development projects and had no alternative future uses.
We estimate our research and development accrued expenses. Our clinical trial accrual process is designed to account for expenses resulting from our obligations under contracts with vendors, consultants and clinical research organizations and clinical site agreements in connection with conducting clinical trials. The financial terms of these contracts are subject to negotiations, which vary from contract to contract and may result in payment flows that do not match the periods over which materials or services are provided to us under such contracts. We account for trial expenses according to the progress of the trial as measured by participant progression and the timing of various aspects of the trial. We determine accrual estimates that take into account discussions with applicable personnel and outside service providers as to the progress or state of completion of trials, or the services completed. During the course of a clinical trial, we adjust our clinical expense recognition if actual results differ from our estimates. We make estimates of our accrued expenses as of each balance sheet date based on the facts and circumstances known to us at that time. Our clinical trial accruals and prepaid assets are dependent upon the timely and accurate reporting of contract research organizations and other third-party vendors.
Stock-Based Compensation. All stock-based payments to employees and to nonemployee directors for their services as directors consisted of grants of restricted stock and stock options, which are measured at fair value on the grant date and recognized in the consolidated statements of operations as compensation expense over the relevant vesting period. In addition, for awards that vest immediately and are nonforfeitable, the measurement date is the date the award is issued.
Derivative Instruments and Warrant Liabilities. The Company evaluates all of its financial instruments, including issued warrants to purchase common stock under ASC 815 – Derivatives and Hedging, to determine if such instruments are derivatives or contain features that qualify as embedded derivatives. For derivative financial instruments that are accounted for as liabilities, the derivative instrument is initially recorded at its fair value and is then re-valued at each reporting date, with changes in the fair value reported in the consolidated statements of operations. The Company uses the Monte Carlo pricing model to value the derivative instruments at inception and subsequent valuation dates, which is adjusted for instrument-specific terms as applicable.
From time to time, certain equity-linked instruments may be classified as derivative liabilities due to the Company having insufficient authorized shares to fully settle the equity-linked financial instruments in shares. In such a case, the Company has adopted a sequencing approach under ASC 815-40, Derivatives and Hedging - Contracts in Entity’s Own Equity to determine the classification of its contracts at issuance and at each subsequent reporting date. If reclassification of contracts between equity and assets or liabilities is necessary, the Company first allocates remaining authorized shares to equity on the basis of the earliest issuance date of potentially dilutive instruments, with the earliest issuance date receiving the first allocation of shares. In the event of identical issuance dates, shares are then allocated to equity beginning with instruments with the latest maturity date first.
Other than contractual obligations incurred in the normal course of business, we do not have any off-balance sheet financing arrangements or liabilities, guarantee contracts, retain or contingent interests in transferred assets or any obligation arising out of a material variable interest in an unconsolidated entity.
97
Recently Adopted Accounting Pronouncements
In December 2023, the FASB issued ASU 2023-09, Improvements to Income Tax Disclosures, which requires entities to disclose disaggregated information about their effective tax rate reconciliations as well as expanded information on income taxes by jurisdiction. The standard is effective for fiscal years beginning after December 15, 2024 on a prospective basis. The Company discloses its income tax rate reconciliation in its annual consolidated financial statements only. The Company adopted the ASU on January 1, 2025 and the impact of the adoption was enhanced disclosure in Note 18.
Recently Issued Accounting Pronouncements
In March 2024, the SEC adopted new rules relating to the disclosure of a range of climate-change-related physical and transition risks, data, and opportunities. The adopted rule contains several new disclosure obligations, including, (i) disclosure on how the board of directors and management oversee climate-related risks and certain climate-related governance items, (ii) disclosure of information related to a registrant’s climate-related targets, goals, and/or transition plans, and (iii) disclosure on whether and how climate-related events and transition activities impact line items above a threshold amount on a registrant’s consolidate financial statements, including the impact of the financial estimates and the assumptions used. This new rule will first be effective in the Company’s disclosures for the year ending December 31, 2027. The Company is in the process of assessing the impact on our consolidated financial statements and disclosures.
In November 2024, the FASB issued ASU 2024-03, Income Statement—Reporting Comprehensive Income—Expense Disaggregation Disclosures, to improve transparency in financial reporting by requiring entities to present more detailed information about the nature of expenses included within the Income Statement. The guidance will first be effective for annual reporting periods beginning after December 15, 2026, and interim reporting periods beginning after December 15, 2027. Early adoption is permitted. The Company is in the process of assessing the impact of ASU 2024-03 on our disclosures.
In December 2025, the FASB issued ASU 2025-10, Government grants – Accounting for Government grants by Business entities, that includes requirements for recognition of government grants in a Company’s financial statements as well as disclosure requirements, including the nature of the government grant received, the accounting policies used to account for the grant, and significant terms and conditions of the grant. The guidance is effective for 2029 interim and annual reporting on a modified prospective, modified retrospective or retrospective approach. Early adoption is permitted as of the beginning of an annual reporting period. The Company is currently evaluating the impact of adoption on its consolidated financial statements.
ITEM 7A – QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Not required under Regulation S-K for “smaller reporting companies.”
98
ITEM 8 – FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
TONIX PHARMACEUTICALS HOLDING CORP.
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Tonix Pharmaceuticals Holding Corp.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Tonix Pharmaceuticals Holding Corp. and Subsidiaries (the “Company”) as of December 31, 2025 and 2024, and the related consolidated statements of operations, comprehensive loss, stockholders’ equity, and cash flows for each of the years then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the consolidated financial position of the Company as of December 31, 2025 and 2024, and the consolidated results of their operations and their cash flows for each of the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has continuing losses and negative cash flows from operating activities that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates relate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of the critical audit matter does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
F-2
Accrued and prepaid balance for research and development expenses
As described in Note 2 to the financial statements, at each balance sheet date the Company estimates its expenses resulting from its obligations under contracts with vendors, clinical research organizations, and consultants in connection with performing pre-clinical and clinical work in preparation for and related to clinical trials. The Company accounts for research and development expenses based on services that have been performed on the Company’s behalf and estimating the level of service performed and the associated costs incurred for the services when an invoice has not been received. The Company’s accrual for pre-clinical and clinical trial expenses of approximately $3.4 million is included in accrued expenses and other current liabilities on the December 31, 2025 consolidated balance sheet. The Company also recorded prepaid pre-clinical and clinical trial expenses of approximately $3.5 million within prepaid expenses and other on the December 31, 2025 consolidated balance sheet. The amounts recorded for clinical trial expenses represent the Company’s estimates of the unpaid and prepaid clinical trial expenses based on facts and circumstances known to the Company at that time and are dependent upon the timely and accurate reporting of contract research organizations and other third-party vendors. The estimation of clinical trial expenses was also identified as acritical accounting estimate by management.
We identified the accrual for research and development expenses as a critical audit matter due to the significant judgment and estimation required by management in determining progress or state of completion of clinical trials or services completed. This in turn led to a high degree of auditor subjectivity, and significant audit effort was required in performing our procedures and evaluating audit evidence relating to estimates made by management.
Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our overall opinion on the financial statements. We obtained an understanding of Management’s process and evaluated the design of controls over developing its estimate of accrued and prepaid research and development expenses, including the process of estimating the expenses incurred to date based on the status of the pre-clinical and clinical trial work. Our procedures also included, among others, reading agreements and contract amendments with vendors, consultants and clinical research organizations and clinical site agreements in connection with conducting clinical trials, and evaluating the significant assumptions described above and the methods used in developing the clinical trial estimates and re-calculating the amounts that were unpaid and prepaid at the balance sheet date. We confirmed certain contractual commitments for completeness of the contract listing, as well as confirmation of work completed, paid and unpaid, directly with the third parties involved in performing the pre-clinical and clinical trial services on behalf of the Company. We also made direct inquiries of Company financial personnel regarding the contract amount including change orders, status and progress to completion of clinical trials, amounts paid to date under each contract, and description of future commitments. We also assessed the historical accuracy of management’s estimates by comparing work completed in the current period to work completed in prior-period to identify unusual fluctuations, if any.
We have served as the Company’s auditor since 2010.
EISNERAMPER LLP
Iselin, New Jersey
March 12, 2026
F-3
TONIX
PHARMACEUTICALS HOLDING CORP.
CONSOLIDATED BALANCE SHEETS
(In Thousands, Except Par Value and Share Amounts)
| December 31, | December 31, | |||||||
| 2025 | 2024 | |||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | $ | ||||||
| Accounts receivable, net | ||||||||
| Inventory | ||||||||
| Prepaid expenses and other current assets | ||||||||
| Total current assets | ||||||||
| Property and equipment, net | ||||||||
| Intangible assets, net | ||||||||
| Operating lease right-to-use assets | ||||||||
| Other non-current assets | ||||||||
| Total assets | $ | $ | ||||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | $ | ||||||
| Accrued expenses and other current liabilities | ||||||||
| Term loan payable, short term | ||||||||
| Lease liability, short term | ||||||||
| Total current liabilities | ||||||||
| Term loan payable, long term | ||||||||
| Lease liability, long term | ||||||||
| Total liabilities | ||||||||
| Commitments (See Note 17) | ||||||||
| Stockholders’ equity: | ||||||||
| Preferred stock, $ par value; shares authorized, shares designated as of both December 31, 2025 and 2024; shares issued and outstanding - as of both December 31, 2025 and 2024 | ||||||||
| Common stock, $ par value; shares authorized; and shares issued and outstanding as of December 31, 2025 and 2024, respectively and shares to be issued as of December 31, 2025 | ||||||||
| Additional paid in capital | ||||||||
| Accumulated deficit | ( |
) | ( |
) | ||||
| Accumulated other comprehensive loss | ( |
) | ( |
) | ||||
| Total stockholders’ equity | ||||||||
| Total liabilities and stockholders’ equity | $ | $ | ||||||
See the accompanying notes to the consolidated financial statements
F-4
TONIX PHARMACEUTICALS HOLDING CORP.
CONSOLIDATED STATEMENTS OF OPERATIONS
(In Thousands, Except Share and Per Share Amounts)
| Year ended December 31, | ||||||||
| 2025 | 2024 | |||||||
| REVENUES: | ||||||||
| Product revenue, net | $ | $ | ||||||
| COSTS AND EXPENSES: | ||||||||
| Cost of sales | ||||||||
| Research and development | ||||||||
| Selling, general and administrative | ||||||||
| Asset impairment charges | ||||||||
| Total Operating Expenses | ||||||||
| Operating loss | ( |
) | ( |
) | ||||
| Grant income | ||||||||
| Gain on change in fair value of warrant liabilities | ||||||||
| Loss on extinguishment of debt | ( |
|||||||
| Interest income | ||||||||
| Interest expense | ( |
) | ( |
) | ||||
| Other expense, net | ( |
) | ( |
) | ||||
| Net loss available to common stockholders | $ | ( |
) | $ | ( |
) | ||
| Net loss to common stockholders per common share, basic and diluted | $ | ) | $ | ) | ||||
| Weighted average common shares outstanding, basic and diluted | ||||||||
See the accompanying notes to the consolidated financial statements
F-5
TONIX PHARMACEUTICALS HOLDING CORP.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
(In Thousands)
| Year ended December 31, | ||||||||
| 2025 | 2024 | |||||||
| Net loss | $ | ( |
) | $ | ( |
) | ||
| Other comprehensive loss: | ||||||||
| Foreign currency translation loss | ( |
) | ( |
) | ||||
| Comprehensive loss | $ | ( |
) | $ | ( |
) | ||
See the accompanying notes to the consolidated financial statements
F-6
TONIX PHARMACEUTICALS HOLDING CORP.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(In Thousands, Except Share and Per Share Amounts)
| Accumulated | ||||||||||||||||||||||||
| Additional | Other | |||||||||||||||||||||||
| Common stock | Paid in | Comprehensive | Accumulated | |||||||||||||||||||||
| Shares | Amount | Capital | Loss | Deficit | Total | |||||||||||||||||||
| Balance, December 31, 2024 | $ | $ | $ | ( |
) | $ | ( |
) | $ | |||||||||||||||
| Repurchase
of common stock under Share Repurchase Program, including transactional expenses of $ |
( |
) | ( |
) | ( |
) | ||||||||||||||||||
| Issuance of commitment shares | ||||||||||||||||||||||||
| Issuance
of common stock under At-the-market offering, net of transactional expenses of $ |
||||||||||||||||||||||||
| Issuance
of common stock and warrants under registered direct Financing, net of transactional expenses of $ |
||||||||||||||||||||||||
| Stock-based compensation | — | |||||||||||||||||||||||
| Foreign currency transaction gain | — | ( |
) | ( |
) | |||||||||||||||||||
| Net loss | — | ( |
) | ( |
) | |||||||||||||||||||
| Balance, December 31, 2025 | $ | $ | $ | ( |
) | $ | ( |
) | $ | |||||||||||||||
See the accompanying notes to the consolidated financial statements
F-7
TONIX PHARMACEUTICALS HOLDING CORP.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(In Thousands, Except Share and Per Share Amounts)
| Accumulated | ||||||||||||||||||||||||
| Additional | Other | |||||||||||||||||||||||
| Common stock | Paid in | Comprehensive | Accumulated | |||||||||||||||||||||
| Shares | Amount | Capital | Loss | Deficit | Total | |||||||||||||||||||
| Balance, December 31, 2023 | $ | $ | $ | ( |
) | $ | ( |
) | $ | |||||||||||||||
| Issuance of common stock upon exercise of prefunded warrants | ||||||||||||||||||||||||
| Issuance
of common stock under At-the-market offering, net of transactional expenses of $ |
||||||||||||||||||||||||
| Issuance
of common stock, net of transactional expenses of $ |
||||||||||||||||||||||||
| Fair value of warrants reclassified from liabilities to equity | — | |||||||||||||||||||||||
| Fair value of warrants classified from equity to liabilities | — | ( |
) | ( |
) | |||||||||||||||||||
| Employee stock purchase plan | ||||||||||||||||||||||||
| Stock-based compensation | — | |||||||||||||||||||||||
| Foreign currency transaction gain | — | ( |
) | ( |
) | |||||||||||||||||||
| Net loss | — | ( |
) | ( |
) | |||||||||||||||||||
| Balance, December 31, 2024 | $ | $ | $ | ( |
) | $ | ( |
) | $ | |||||||||||||||
See the accompanying notes to the consolidated financial statements
F-8
TONIX PHARMACEUTICALS HOLDING CORP.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In Thousands)
| Year ended December 31, | ||||||||
| 2025 | 2024 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||
| Net loss | $ | ( |
) | $ | ( |
) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation and amortization | ||||||||
| Bad debt expense | ||||||||
| Asset impairment charges | ||||||||
| Stock-based compensation | ||||||||
| Change in fair value of warrant liabilities | ( |
) | ||||||
| Loss on extinguishment of debt | ||||||||
| Loss on note | ||||||||
| Inventory write-off | ||||||||
| Amortization of debt discounts | ||||||||
| Issuance costs of derivative instrument | ||||||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts receivable | ( |
) | ( |
) | ||||
| Inventory | ||||||||
| Prepaid expenses and other | ( |
|||||||
| Accounts payable | ||||||||
| Operating lease liabilities and ROU asset, net | ( |
) | ( |
) | ||||
| Accrued expenses and other current liabilities | ||||||||
| Net cash used in operating activities | ( |
) | ( |
) | ||||
| CASH FLOWS FROM INVESTING ACTIVITIES: | ||||||||
| Issuance of note | ( |
|||||||
| Proceeds from note | ||||||||
| Purchase of property and equipment | ( |
) | ( |
) | ||||
| Net cash used in investing activities | ( |
) | ( |
) | ||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||
| Deferred payment related to purchase of business | ( |
) | ||||||
| Proceeds from ESPP | ||||||||
| Repayments of term loan | ( |
) | ( |
) | ||||
| Proceeds,
net of $ |
||||||||
| Repurchase
of common stock, net of $ |
( |
|||||||
| Net cash provided by financing activities | ||||||||
| Effect of currency rate change on cash | ( |
|||||||
| Net increase in cash, cash equivalents and restricted cash | ||||||||
| Cash, cash equivalents and restricted cash beginning of the period | ||||||||
| Cash, cash equivalents and restricted cash end of period | $ | $ | ||||||
| Supplemental disclosures of cash flow information: | ||||||||
| Interest paid | $ | $ | ||||||
| Non cash investing and financing activity: | ||||||||
| Purchases of property and equipment included in accounts payable and accrued liabilities | $ | $ | ||||||
| Net ATM proceeds received after year-end | $ | $ | ||||||
| Issuance costs from derivative instruments | $ | |||||||
| New operating leases and lease amendments | $ | |||||||
See the accompanying notes to consolidated financial statement
F-9
TONIX PHARMACEUTICALS HOLDING CORP.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1 – BUSINESS
Tonix Pharmaceuticals (“Tonix” or the “Company”) is a fully-integrated biopharmaceutical company commercializing and developing innovative therapies for central nervous system (“CNS”) disorders, immunology, infectious diseases, and rare diseases. Our portfolio consists of both commercial and development-stage programs.
In August 2025, Tonix received approval from the U.S. Food and Drug Administration (“FDA”) for TONMYA™ (cyclobenzaprine HCl sublingual tablets) for the treatment of fibromyalgia. TONMYA, Tonix’s first internally developed product to become FDA approved, was commercially launched by the Company in the United States on November 17, 2025. TONMYA is the first new medicine for fibromyalgia in more than 15 years and is a centrally acting, non-opioid analgesic designed for bedtime administration and long-term use. The approval and launch of TONMYA marked major milestones in Tonix’s evolution. The Company holds worldwide commercialization rights to TONMYA. In addition to TONMYA, Tonix markets two FDA-approved prescription products for the treatment of acute migraine: Zembrace® SymTouch® (sumatriptan injection) and Tosymra® (sumatriptan nasal spray). Tonix’s commercial platform includes sales, marketing, market access, distribution, and patient support capabilities.
Tonix maintains a diversified development pipeline generated through internal discovery, in-licensing, acquisitions, and collaborations with academic and non-profit institutions. The Company’s pipeline addresses conditions that span central nervous system (“CNS”), infectious disease, immunology, and rare disease, with multiple programs in clinical and preclinical development. The proprietary cyclobenzaprine HCl sublingual tablet formulation contained in TONMYA is referred to as “TNX-102 SL” outside of the fibromyalgia indication. Tonix is exploring the utility of TNX-102 SL (sublingual cyclobenzaprine) in Phase 2 clinical trials for major depressive disorder and acute stress disorder. TNX-102 SL is being developed to treat acute stress reaction and acute stress disorder under an Investigator-Initiated investigational new drug application (“IND”) at the University of North Carolina in the ongoing OASIS study funded by the U.S. Department of Defense (“DoD”). A Phase 2 study of TNX-102 SL for major depressive disorder is expected to commence mid-2026 under a Tonix IND that has been cleared by FDA.
Tonix’s clinical stage infectious disease portfolio includes monoclonal antibody TNX-4800 (anti-OspA from Borrelia burgdorferi) for seasonal prevention of Lyme disease, for which initiation of a Phase 2 field study is planned for the first half of 2027 and a Phase 2 human challenge study is planned for 2028, pending FDA clearances.
Tonix’s clinical-stage immunology development portfolio consists of biologics to address organ transplant rejection, autoimmunity and cancer, including TNX-1500, which is a Phase 2- ready Fc-modified humanized monoclonal antibody targeting CD40-ligand (CD40L or CD154) being developed for the prevention of allograft rejection and for the treatment of autoimmune diseases.
Another CNS candidate in clinical development is TNX-1300 (double-mutant cocaine esterase), which is in Phase 2 for the treatment of cocaine intoxication. TNX-1300 has been granted Breakthrough Therapy designation by the FDA.
Tonix’s clinical-stage rare disease portfolio includes TNX-2900, intranasal oxytocin potentiated with magnesium, in development for Prader-Willi syndrome and expected to start a Phase 2 study in the first quarter of 2027.
Tonix’s pre-clinical, pre-IND infectious disease portfolio includes TNX-801 (horsepox, live virus vaccine), as vaccine for mpox and smallpox. We own a facility in Dartmouth, MA that was purpose-built to manufacture TNX-801 under Good Manufacturing Practices (GMP) to support clinical development and potential commercialization.
Tonix’s pre-IND infectious disease portfolio also includes TNX-4200, which is a small molecule broad-spectrum antiviral agent targeting CD45 for the prevention or treatment of high lethality infections to improve the medical readiness of military personnel in biological threat environments. The TNX-4200 program is supported by a $34 million contract over five years from the U.S. DoD’s Defense Threat Reduction Agency (DTRA). Tonix owns and operates a state-of-the art research facility in Frederick, Maryland that supports this research.
Tonix’s pre-IND pre-clinical immunology portfolio includes TNX-1700, which is a fusion protein of TFF2 and albumin is in preclinical development for the treatment of gastric cancer in combination with PD-1 blockade in collaboration with Columbia University.
Tonix’s pre-clinical, pre-IND CNS portfolio also includes TNX-4900, a highly selective small-molecule Sigma-1 receptor (“S1R”) antagonist for neuropathic pain.
The consolidated financial statements include the accounts of Tonix Pharmaceuticals Holding Corp. and its wholly owned subsidiaries, Tonix Sub, Krele LLC, Tonix Pharmaceuticals (Canada), Inc., Tonix Medicines, Jenner Institute LLC, Tonix R&D Center LLC, Tonix Pharma Holdings Limited and Tonix Pharma Limited (collectively, the “Company” or “Tonix”). All intercompany balances and transactions have been eliminated in consolidation.
Going Concern
The
accompanying financial statements have been prepared on a basis which assumes that the Company will continue as a going concern
and which contemplates the realization of assets and satisfaction of liabilities and commitments in the normal course of business.
The Company has suffered recurring losses from operations and negative cash flows from operating activities. At December 31, 2025,
the Company had working capital of approximately $
F-10
The Company believes that its cash resources at December 31, 2025, and
the net proceeds of $
These factors raise substantial doubt about the Company’s ability to continue as a going concern. The Company continues to face significant challenges and uncertainties and must successfully launch TONMYA and obtain additional funding through public and private financing and collaborative arrangements with strategic partners to increase the funds available to fund operations. However, the Company may not be able to raise capital on terms acceptable to the Company, or at all. Without the successful product launch of TONMYA and obtaining additional funds, the Company may be forced to delay, scale back or eliminate some or all of its research and development activities or other operations, and potentially delay product development in an effort to maintain sufficient funds to continue operations. If any of these events occurs, the Company’s ability to achieve development and commercialization goals will be adversely affected and the Company may be forced to cease operations. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
NOTE 2 – SIGNIFICANT ACCOUNTING POLICIES
Reverse Stock Split
On
February 5, 2025, the Company effected a
Risks and uncertainties
The Company’s primary efforts are devoted to commercializing its approved products and conducting research and development of innovative pharmaceutical and biological products to address public health challenges. The Company has experienced net losses and negative cash flows from operations since inception and expects these conditions to continue for the foreseeable future. Further, the Company currently generates revenue from the sale of its commercial products, TONMYA, Zembrace SymTouch and Tosymra. There is no assurance that the Company will be able to generate sufficient cash flow to fund operations from the sale of its commercial products or products in development, if and when approved. In addition, there can be no assurance that the Company’s research and development will be successfully completed or that any product in development will be approved or commercially viable.
Use of estimates
The preparation of financial statements in accordance with Generally Accepted Accounting Principles (“GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosures of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates. Significant estimates include, but are not limited to, impairments, provisions for product returns, coupons, rebates, chargebacks, discounts, allowances, inventory realization, the assumptions used in the fair value of stock-based compensation and other equity instruments, and the percent of completion of research and development contracts.
Segment Information and Concentrations
Operating
segments are defined as components of an enterprise about which separate discrete information is available for evaluation by the
chief operating decision maker (“CODM”), or decision-making group, in deciding how to allocate resources and in assessing
performance. The Company considers its chief executive officer to be the Company’s CODM. The CODM manages its operations
and allocates resources based on the Company’s consolidated results and therefore operates as
Segment revenue, profit or loss, significant segment expenses and other segment items - The accounting policies of the Company’s single operating and reportable segment are the same as those described in the summary of significant accounting policies. The Company’s method for measuring segment profitability includes net income (loss), which the CODM uses to assess performance and make decisions for resource allocation, consistent with the measurement principals for net income (loss) as reported on the Company’s consolidated statements of operations. The significant expenses regularly reviewed by the CODM are consistent with those reported on the Company’s consolidated statements of operations, and expenses are not regularly reviewed on a more disaggregated basis for purposes of assessing segment performance and deciding how to allocate resources.
F-11
The
Company has three products that accounted for $ million, representing
As
of December 31, 2025, accounts receivable from three customers accounted for
For
the year ended December 31, 2025, revenues from five customers accounted for
Cash, Cash Equivalents and Restricted Cash
The
Company considers cash equivalents to be those investments which are highly liquid, readily convertible to cash and have an original
maturity of three months or less when purchased. At December 31, 2025, and 2024, cash equivalents, which consisted of money market
funds and other cash equivalents, amounted to approximately $
The following table provides a reconciliation of cash, cash equivalents and restricted cash reported within the consolidated balance sheets that sum to the total of the same amounts shown in the consolidated statement of cash flows:
| December
31, 2025 |
December
31, 2024 |
|||||||
| (in thousands) | ||||||||
| Cash and cash equivalents | $ | $ | ||||||
| Restricted cash | ||||||||
| Total | $ | $ | ||||||
Accounts Receivable, net
Accounts receivable consists of amounts due from our wholesale and other third-party distributors and pharmacies and have standard payment terms that generally require payment within 30 to 90 days. For certain customers, the accounts receivable for the customer is net of cash discounts, chargebacks and customer rebates. We do not adjust our receivables for the effects of a significant financing component at contract inception if we expect to collect the receivables in one year or less from the time of sale. We provide reserves against accounts receivable for estimated losses that may result from a customer’s inability to pay. Amounts determined to be uncollectible are charged or written-off against the reserve.
As
of December 31, 2025 and 2024, the Company had $
Concentration of Credit Risk
Financial instruments that potentially subject us to concentrations of credit risk include cash and cash equivalents, and receivables. We attempt to minimize the risks related to cash and cash equivalents by investing in money market accounts, and we have established guidelines related to credit ratings and maturities intended to safeguard principal balances and maintain liquidity.
F-12
TONIX PHARMACEUTICALS HOLDING CORP.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Concentrations of credit risk with respect to receivables, which are typically unsecured, are somewhat mitigated due to the variety of customers using our products, as well as their dispersion across different geographic areas.
We monitor the financial performance and creditworthiness of our customers so that we can properly assess and respond to changes in their credit profile. We continue to monitor these conditions and assess their possible impact on our business.
Inventories
Inventories
are recorded at the lower of cost or net realizable value, with cost determined by the weighted average cost method. Acquired
inventory was valued at estimated selling price less a reasonable margin. The Company periodically reviews the composition of
inventory in order to identify excess, obsolete, slow-moving or otherwise non-saleable items taking into account anticipated future
sales compared with quantities on hand, and the remaining shelf life of goods on hand. If non-saleable items are observed and
there are no alternate uses for the inventory, the Company records a write-down to net realizable value in the period that the
decline in value is first recognized. During the year ended December 31, 2025 and 2024, the Company recorded write-downs related
to Tosymra and Zembrace finished goods inventory of approximately $
Property and equipment
Property
and equipment are stated at cost, less accumulated depreciation. Depreciation and amortization is calculated using the straight-line
method over the asset’s estimated useful life, which ranges from
Intangible assets, net
Intangible
assets deemed to have finite lives are carried at acquisition-date fair value less accumulated amortization and impairment, if
any. Finite-lived intangible assets consisted of developed technology intangible assets acquired in connection with the acquisition
of certain products from Upsher Smith Laboratories, LLC (“Upsher Smith”) consummated on June 30, 2023 (See Note 5).
The acquired intangible assets were amortized using the straight-line method over the estimated useful lives of the respective
assets. Amortization expense the year ended December 31, 2024, was $
Impairment testing of long-lived assets
The Company evaluates long-lived assets for impairment, including property and equipment, finite-lived intangibles assets and operating lease right-to-use assets whenever events or changes in circumstances indicate that their net book value may not be recoverable. When such factors and circumstances exist, the Company compares the projected undiscounted future cash flows associated with the related asset or group of assets over their estimated useful lives against their respective carrying amount. Impairment, if any, is based on the excess of the carrying amount over the fair value, based on market value when available, or discounted expected cash flows, of those assets and is recorded in the period in which the determination is made. For the year ended December 31, 2025, the Company concluded that no impairment existed.
During
the second quarter of 2024, the Company identified certain triggering events related to its decommissioned Advance Development
Center in Dartmouth, Massachusetts (“ADC”). The Company determined that the carrying value of the ADC was not recoverable
and that the carrying value exceeded its fair value. As such, the Company recorded a non-cash impairment charge of $
F-13
TONIX PHARMACEUTICALS HOLDING CORP.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Additionally,
due to a sustained decline in revenues and continued delays in building out the sales team for its commercialized products, the
Company also tested its commercialized products asset group for recoverability during the second quarter of 2024. The Company
determined that the carrying value was not recoverable and therefore estimated the fair value of the asset group using a discounted
cash flow analysis. As the carrying value of the asset group significantly exceeded its fair value, the Company recorded a full
non-cash impairment charge of $
Goodwill
Goodwill
represents the excess of the aggregate purchase price over the fair value of the net tangible and intangible assets acquired in
a business combination. Goodwill is reviewed for impairment on an annual basis, or more frequently if events or changes in circumstances
indicate that the carrying amount of goodwill may be impaired. The Company previously recognized goodwill in connection with the
USL Acquisition consummated on June 30, 2023 (See Note 5). The Company completed the required annual impairment test for goodwill
during the second quarter of 2024, which resulted in full non-cash impairment of the Company’s $
Leases
The Company determines if an arrangement is, or contains, a lease at inception. Operating leases are included in operating lease right-of-use (“ROU”) assets, operating lease liabilities, current and operating lease liabilities, noncurrent in the Company’s consolidated balance sheets. ROU assets represent the Company’s right to use an underlying asset for the lease term and lease liabilities represent its obligation to make lease payments arising from the lease. Operating lease ROU assets and liabilities are recognized at commencement date based on the present value of lease payments over the lease term. As the Company’s leases do not provide an implicit rate, the Company uses an incremental borrowing rate based on the information available at the transition date and subsequent lease commencement dates in determining the present value of lease payments. This is the rate the Company would have to pay if borrowing on a collateralized basis over a similar term to each lease. The operating lease ROU asset excludes lease incentives. The Company’s lease terms may include options to extend or terminate the lease when it is reasonably certain that the Company will exercise that option. Lease expense for lease payments made under operating leases is recognized on a straight-line basis over the lease term.
Revenue Recognition
The
Company records and recognizes revenue in a manner that depicts the transfer of promised goods or services to customers in an
amount that reflects the consideration to which the Company expects to be entitled in exchange for those goods or services. The
Company’s revenues primarily result from contracts with customers, which are generally short-term and have a single performance
obligation - the delivery of product. The Company’s performance obligation to deliver products is satisfied at the point
in time that the goods are received by the customer, which is when the customer obtains title to and has the risks and rewards
of ownership of the products, which is generally upon shipment or delivery to the customer as stipulated by the terms of the sale
agreements. The transaction price is the amount of consideration to which the Company expects to be entitled in exchange for transferring
promised goods to a customer. The consideration promised in a contract with a customer may include fixed amounts, variable amounts,
or both. Our contractual payment terms are typically
Revenues from product sales, net of gross-to-net deductions, are recorded only to the extent a significant reversal in the amount of cumulative revenue recognized is not probable of occurring and when the uncertainty associated with gross-to-net deductions is subsequently resolved. Taxes assessed by governmental authorities and collected from customers are excluded from product sales. Shipping and handling activities are considered to be fulfillment activities and not a separate performance obligation.
F-14
Many of the Company’s products sold are subject to a variety of deductions. Revenues are recognized net of estimated rebates and chargebacks, cash discounts, distributor fees, sales return provisions and other related deductions. Deductions to product sales are referred to as gross-to-net deductions and are estimated and recorded in the period in which the related product sales occur. Accruals for these provisions are presented in the consolidated financial statements as reductions to gross sales in determining net sales, and as a contra asset within accounts receivable, net (if settled via credit) and other current liabilities (if paid in cash). Amounts recorded for revenue deductions can result from a complex series of judgements about future events and uncertainties and can rely heavily on estimates and assumptions. The following section briefly describes the nature of the Company’s provisions for variable consideration and how such provisions are estimated:
Chargebacks - The Company sells a portion of its products indirectly through wholesaler distributors, and enters into specific agreements with these indirect customers to establish pricing for the Company’s products, and in-turn, the indirect customers and entities independently purchase these products. Because the price paid by the indirect customers and/or entities is lower than the price paid by the wholesaler, the Company provides a credit, called a chargeback, to the wholesaler for the difference between the contractual price with the indirect customers and the wholesale customer’s purchase price. The Company’s provision for chargebacks is based on expected sell-through levels by the Company’s wholesale customers to the indirect customers and estimated wholesaler inventory levels as well as historical chargeback rates. The Company continually monitors its reserve for chargebacks and adjusts the reserve accordingly when expected chargebacks differ from actual experience.
Rebates - The Company participates in certain government and specific sales rebate programs which provides discounted prescription drugs to qualified recipients, and primarily relate to Medicaid and managed care rebates in the U.S., pharmacy rebates, Tri-Care rebates and discounts, specialty pharmacy program fees and other governmental rebates or applicable allowances.
| ● | Managed Care Rebates are processed in the quarter following the quarter in which they are earned. The managed care reporting entity submits utilization data after the end of the quarter and the Company processes the payment in accordance with contract terms. All rebates earned but not paid are estimated by the Company according to historical payments trended for market growth assumptions. |
| ● | Medicaid and State Agency rebates are based upon historical experience of claims submitted by various states. The Company monitors Medicaid legislative changes to determine what impact such legislation may have on the provision for Medicaid rebates. The accrual of State Agency reserves is based on historical payment rates. There is an approximate three-month lag from the time of product sale until the rebate is paid. |
| ● | Tri-Care represents a regionally managed health care program for active duty and retired members, dependents and survivors of the US military. The Tri-Care program supplements health care resources of the US military with civilian health care professionals for greater access and quality healthcare coverage. Through the Tri-Care program, the Company provides pharmaceuticals on a direct customer basis. Prices of pharmaceuticals sold under the Tri-Care program are pre-negotiated and a reserve amount is established to represent the proportionate rebate amount associated with product sales. |
| ● | Coverage Gap refers to the Medicare prescription drug program and represents specifically the period between the initial Medicare Part D prescription drug program coverage limit and the catastrophic coverage threshold. Applicable pharmaceutical products sold during this coverage gap timeframe are discounted by the Company. Since the nature of the program is that coverage limits are reset at the beginning of the calendar year; the payments escalate each quarter as the participants reach the coverage limit before reaching the catastrophic coverage threshold. The Company has determined that the cost of this reserve will be viewed as an annual cost. Therefore, the accrual will be incurred evenly during the year with quarterly review of the liability based on payment trends and any revision to the projected annual cost. |
Prompt-Pay and other Sales Discounts - The Company provides for prompt pay discounts, which early payments are recorded as a reduction of revenue and as a reduction in the accounts receivable at the time of sale based on the customer’s contracted discount rate. Consumer sales discounts represent programs the Company has in place to reduce costs to the patient. This includes copay buy down and eVoucher programs.
Product Returns - Consistent with industry practice, the Company offers customers a right to return any unused product. The customer’s right of return commences typically six months prior to product expiration date and ends one year after product expiration date. Products returned for expiration are reimbursed at current wholesale acquisition cost or indirect contract price. The Company estimates the amount of its product sales that may be returned by the Company’s customers and accrues this estimate as a reduction of revenue in the period the related product revenue is recognized. For Zembrace and Tosymra, the Company estimates product returns as a percentage of sales to its customers. The rate is estimated by using historical sales information, including its visibility and estimates into the inventory remaining in the distribution channel. For TONMYA, until sufficient historical information is available, the rate is based on the industry average for a similar product. Adjustments are made to the current provision for returns when data suggests product returns may differ from original estimates.
F-15
TONIX PHARMACEUTICALS HOLDING CORP.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Research and Development Costs
The Company outsources certain of its research and development efforts and expenses these costs as incurred, including the cost of manufacturing products for testing, as well as licensing fees and costs associated with planning and conducting clinical trials. The value ascribed to patents and other intellectual property acquired has been expensed as research and development costs, as such property is related to particular research and development projects and had no alternative future uses.
The Company estimates its expenses resulting from its obligations under contracts with vendors, clinical research organizations and consultants and under clinical site agreements in connection with conducting clinical trials.
The financial terms of these contracts are subject to negotiations, which vary from contract to contract and may result in payment flows that do not match the periods over which materials or services are provided under such contracts. The Company accounts for trial expenses according to the timing of various aspects of the trial. The Company determines accrual estimates taking into account discussion with applicable personnel and outside service providers as to the progress or state of consummation of trials, or the services completed.
During the course of a clinical trial, the Company adjusts its clinical expense recognition if actual results differ from its estimates. The Company makes estimates of its accrued expenses as of each balance sheet date based on the facts and circumstances known to it at that time. The Company’s clinical trial accruals are dependent upon the timely and accurate reporting of contract research organizations and other third-party vendors.
Advertising and Promotion Costs
Advertising
and promotion costs are expensed as incurred. The Company recorded advertising and promotion expenses of approximately
$
Government Grants
From time to time, the Company may enter into arrangements with governmental entities for the purpose of obtaining funding for research and development activities. The Company is reimbursed for costs incurred that are associated with specified research and development activities included in the grant application approved by the government authority and, in certain arrangements. U.S. GAAP does not have specific accounting standards covering government grants to business entities. The Company applies International Accounting Standards 20 (“IAS 20”), Accounting for Government Grants and Disclosure of Government Assistance by analogy when accounting for government grants.
Under IAS 20, government grants are initially recognized when there is reasonable assurance the conditions of the grant will be met and the grant will be received. After initial recognition, government grants received are recognized in earnings in the same period the underlying costs for which the grant is intended to compensate are incurred. The Company classifies government grants received under these arrangements as either a reduction to the related research and development expense or as grant income in the consolidated statements of operations, depending on the fee structure of the arrangement. The Company also applies the disclosure requirements of ASC 832, Government Assistance.
In
August 2022, the Company received a Cooperative Agreement grant from the National Institute on Drug Abuse (“NIDA”),
part of the National Institutes of Health, to support the development of its TNX-1300 product candidate for the treatment of cocaine
intoxication. During the year ended December 31, 2025, the Company recorded $
In
June 2024, the Company was awarded a prototype Other Transaction Agreement from the Defense Threat Reduction Agency (“DTRA”),
an agency within the U.S. Department of Defense, to fund the Company’s TNX-4200 program for the development of a small molecule
broad-spectrum antiviral for the prevention or treatment of viral infections to improve the medical readiness of military personnel
in biological threat environments. The DTRA grant provides for payments totaling up to $
All stock-based payments to employees and to nonemployees for their services, including grants of restricted stock units (“RSUs”), and stock options, are measured at fair value on the grant date and recognized in the consolidated statements of operations as compensation expense over the requisite service period. The Company accounts for share-based awards in accordance with the provisions of the Accounting Standards Codification (“ASC”) 718, Compensation – Stock Compensation.
F-16
TONIX PHARMACEUTICALS HOLDING CORP.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Foreign Currency Translation
Operations of the Company’s Canadian subsidiary, Tonix Pharmaceuticals (Canada), Inc., are conducted in local currency, which represents its functional currency. The U.S. dollar is the functional currency of the other foreign subsidiaries.
Balance sheet accounts of the Canadian subsidiary were translated from foreign currency into U.S. dollars at the exchange rate in effect at the balance sheet date and income statement accounts were translated at the average rate of exchange prevailing during the period. Translation adjustments resulting from this process were included in accumulated other comprehensive loss on the consolidated balance sheets.
Comprehensive Income (Loss)
Comprehensive income (loss) is defined as the change in equity of a business during a period from transactions and other events and circumstances from non-owners sources. It includes all changes in equity during a period except those resulting from investments by owners and distributions to owners. Other comprehensive income (loss) represents foreign currency translation adjustments.
Income Taxes
Deferred income tax assets and liabilities are determined based on the estimated future tax effects of net operating loss and credit carryforwards and temporary differences between the tax basis of assets and liabilities and their respective financial reporting amounts measured at the current enacted tax rates. The Company records a valuation allowance on its deferred income tax assets if it is not more likely than not that these deferred income tax assets will be realized.
The Company recognizes a tax benefit from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by taxing authorities, based on the technical merits of the position. The tax benefits recognized in the consolidated financial statements from such a position are measured based on the largest benefit that has a greater than 50% likelihood of being realized upon ultimate settlement. As of December 31, 2025, the Company has not recorded any unrecognized tax benefits. The Company’s policy is to recognize interest and penalties accrued on any unrecognized tax benefits as a component of income tax expense.
On July 4, 2025, the One Big Beautiful Bill Act (“OBBBA”) was enacted in the United States. The OBBBA includes significant provisions, such as the permanent extension of certain expiring provisions of the Tax Cuts and Jobs Act, and the restoration of favorable tax treatment for certain business provisions. The legislation has multiple effective dates, with certain provisions effective in 2025 and others implemented through 2027. The Company assessed the changes and concluded that they were not material.
Derivative Instruments and Warrant Liabilities
The Company evaluates all of its financial instruments, including issued warrants to purchase common stock under ASC 815 – Derivatives and Hedging, to determine if such instruments are derivatives or contain features that qualify as embedded derivatives (See Note 13). For derivative financial instruments that are accounted for as liabilities, the derivative instrument is initially recorded at its fair value and is then re-valued at each reporting date, with changes in the fair value reported in the consolidated statements of operations. The Company uses the Black-Scholes option pricing model to value the derivative instruments at inception and subsequent valuation dates, which is adjusted for instrument-specific terms as applicable.
From time to time, certain equity-linked instruments may be classified as derivative liabilities due to the variable exercise price of the shares to fully settle the equity-linked financial instruments in shares. In such case, the Company has adopted a sequencing approach under ASC 815-40, Derivatives and Hedging - Contracts in Entity’s Own Equity to determine the classification of its contracts at issuance and at each subsequent reporting date.
In the event that reclassification of contracts between equity and assets or liabilities is necessary, the Company first allocates remaining authorized shares to equity on the basis of the earliest issuance date of potentially dilutive instruments, with the earliest issuance date receiving the first allocation of shares. In the event of identical issuance dates, shares are then allocated to equity beginning with instruments with the latest maturity date first.
F-17
TONIX PHARMACEUTICALS HOLDING CORP.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The
classification of derivative instruments is reassessed at each reporting date. If the classification changes as a result of events
during a reporting period, the instrument is reclassified as of the date of the event that caused the reclassification. There
is no limit on the number of times a contract may be reclassified. During the year ended December 31, 2025, the Company recorded
$
The Company evaluated the contract that includes the right to require Lincoln Park to purchase shares of Common Stock in the future (“purchased put right”) considering the guidance in ASC 815-40, and concluded that it is an equity-linked contract that does not qualify for equity classification, and therefore requires fair value accounting as a derivative asset (liability). The Company has analyzed the terms of the purchased put right and has concluded that it had insignificant value as of December 31, 2025.
Reclassifications
Certain prior year amounts have been reclassified for consistency with the current year presentation. These reclassifications had no effect on the reported results of operations.
The computation of basic and diluted loss per share for the year ended December 31, 2025 and 2024 excludes potentially dilutive securities when their inclusion would be anti-dilutive, or if their exercise prices were greater than the average market price of the common stock during the period. Prefunded warrants are assumed exercised on date of issuance and are included in the basic earnings per share (“EPS”) calculation.
All warrants (See Note 15) issued participate on a one-for-one basis with common stock in the distribution of dividends, if and when declared by the Board of Directors, on the Company’s common stock. For purposes of computing EPS, these warrants are considered to participate with common stock in earnings of the Company. Therefore, the Company calculates basic and diluted EPS using the two-class method. Under the two-class method, net income for the period is allocated between common stockholders and participating securities according to dividends declared and participation rights in undistributed earnings. No income was allocated to the warrants for the year ended December 31, 2025, and 2024, as results of operations were a loss for the periods.
| 2025 | 2024 | |||||||
| Warrants to purchase common stock (excluding prefunded warrants) | ||||||||
| Options to purchase common stock | ||||||||
| Totals | ||||||||
Recently Adopted Accounting Pronouncements
In December 2023, the FASB issued ASU 2023-09, Improvements to Income Tax Disclosures, which requires entities to disclose disaggregated information about their effective tax rate reconciliations as well as expanded information on income taxes by jurisdiction. The standard is effective for fiscal years beginning after December 15, 2024 on a prospective basis. The Company discloses its income tax rate reconciliation in its annual consolidated financial statements only. The Company adopted the ASU on January 1, 2025, and the impact of the adoption was enhanced disclosure in Note 18.
Recently Issued Accounting Pronouncements
In March 2024, the SEC adopted new rules relating to the disclosure of a range of climate-change-related physical and transition risks, data, and opportunities. The adopted rule contains several new disclosure obligations, including, (i) disclosure on how the board of directors and management oversee climate-related risks and certain climate-related governance items, (ii) disclosure of information related to a registrant’s climate-related targets, goals, and/or transition plans, and (iii) disclosure on whether and how climate-related events and transition activities impact line items above a threshold amount on a registrant’s consolidated financial statements, including the impact of the financial estimates and the assumptions used. This new rule will first be effective in the Company’s disclosures for the year ending December 31, 2027. The Company is in the process of assessing the impact on our consolidated financial statements and disclosures.
In November 2024, the FASB issued ASU 2024-03, Income Statement—Reporting Comprehensive Income—Expense Disaggregation Disclosures, to improve transparency in financial reporting by requiring entities to present more detailed information about the nature of expenses included within the Income Statement. The guidance will first be effective for annual reporting periods beginning after December 15, 2026, and interim reporting periods beginning after December 15, 2027. Early adoption is permitted. The Company is in the process of assessing the impact of ASU 2024-03 on our financial statements.
F-18
TONIX PHARMACEUTICALS HOLDING CORP.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
In December 2025, the FASB issued ASU 2025-10, Government grants – Accounting for Government grants by Business entities, that includes requirements for recognition of government grants in a Company’s financial statements as well as disclosure requirements, including the nature of the government grant received, the accounting policies used to account for the grant, and significant terms and conditions of the grant. The guidance is effective for 2029 interim and annual reporting on a modified prospective, modified retrospective or retrospective approach. Early adoption is permitted as of the beginning of an annual reporting period. The Company is currently evaluating the impact of adoption on its consolidated financial statements.
NOTE 3 – INVENTORY
The components of inventory consisted of the following (in thousands):
| December
31, 2025 |
December
31, 2024 |
|||||||
| (in thousands) | ||||||||
| Raw Materials | $ | $ | ||||||
| Work-in-process | ||||||||
| Finished Goods | ||||||||
| Total Inventory | $ | $ | ||||||
NOTE 4 – PROPERTY AND EQUIPMENT, NET
Property and equipment, net consisted of the following (in thousands):
| December
31, 2025 |
December
31, 2024 |
|||||||
| (in thousands) | ||||||||
| Property and equipment, net: | ||||||||
| Land | $ | $ | ||||||
| Land improvements | ||||||||
| Buildings | ||||||||
| Office furniture and equipment | ||||||||
| Laboratory equipment | ||||||||
| Leasehold improvements | ||||||||
| Property and equipment gross | ||||||||
| Less: Accumulated depreciation and amortization | ( |
) | ( |
) | ||||
| Property and equipment, net | $ | $ | ||||||
NOTE 5 – GOODWILL AND INTANGIBLE ASSETS
The following table provides the gross carrying amount and accumulated amortization for each major class of intangible asset:
| December
31, 2025 |
December
31, 2024 |
|||||||
| (in thousands) | ||||||||
| Intangible assets subject to amortization | ||||||||
| Developed technology | $ | $ | ||||||
| Less: Impairment charge | ||||||||
| Less: Accumulated amortization | ||||||||
| Total | $ | $ | ||||||
| Intangible assets not subject to amortization | ||||||||
| Internet domain rights | $ | $ | ||||||
| Total intangible assets, net | $ | $ | ||||||
During
the year ended December 31, 2024, the Company recorded amortization of $
As
a result of certain triggering events identified impacting the Company’s commercialized products asset group during the
second quarter of 2024, the Company tested the asset group for impairment as of June 30, 2024, resulting in a full impairment
of its Zembrace and Tosymra developed technology intangible assets, of $
F-19
TONIX PHARMACEUTICALS HOLDING CORP.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 6 – FAIR VALUE MEASUREMENTS
Fair value measurements affect the Company’s accounting for certain of its financial assets. Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date and is measured according to a hierarchy that includes:
| Level 1: | Observable inputs, such as quoted prices in active markets. |
| Level 2: | Inputs, other than quoted prices in active markets, that are observable either directly or indirectly. Level 2 assets and liabilities include debt securities with quoted market prices that are traded less frequently than exchange-traded instruments. This category includes U.S. government agency-backed debt securities and corporate-debt securities. |
| Level 3: | Unobservable inputs in which there is little or no market data. |
As
of December 31, 2025, and 2024, the Company used Level 1 quoted prices in active markets to value cash equivalents of $
The Company used the Black-Scholes option pricing model to estimate the fair value of the Series D Warrants and the Series C Warrants using significant unobservable inputs, which represent Level 3 measurements within the fair value hierarchy. For periods prior to the receipt of stockholder approval, the fair value was then adjusted by applying a discount for lack of marketability (“DLOM”) based on the expected timing of receipt of stockholder approval to increase the number of authorized shares and to allow the Warrants to become exercisable in accordance with Nasdaq Listing Rule 5635. Additionally, between April 1, 2024 and May 22, 2024, Level 3 liabilities included a portion of the Company’s outstanding August 2023 Warrants, Series A Warrants, Series B Warrants, Series C Warrants, and Series D Warrants (collectively, the “Existing Warrants”), as a result of certain Warrant Amendments entered into upon the closing of an equity financing on April 1, 2024, which provided for adjustments to the exercise prices of the Existing Warrants, contingent on approval by the Company’s stockholders of a proposal to allow the Existing Warrants to become exercisable in accordance with Nasdaq Listing Rule 5635. The Company determined that the exercise price adjustment provision that is contingent on stockholder approval precluded the Existing Warrants from being indexed to the Company’s own stock, and therefore were reclassified to liabilities at post-modification fair value on April 1, 2024. After the Company received stockholder approval on May 22, 2024, thereby reducing the exercise prices of each of the Existing Warrants to $ per share, the Existing Warrants met all requirements for equity classification and the Company reclassified them to equity as of May 22, 2024. To estimate the fair value of the Existing Warrants on the reclassification dates, the Company used a Black-Scholes option pricing model, probability weighted for different scenarios as applicable.
The following table summarizes the range of significant assumptions used in determining the fair value of liability-classified warrants on the respective reclassification dates for the year ended December 31, 2024:
| Year ended | |||||||
| December 31, 2024 | |||||||
| Common stock price | $ | - | |||||
| Risk-free rate | |||||||
| Expected term (in years) | |||||||
| Expected volatility | |||||||
| Discount for lack of marketability | N/A | ||||||
For the year ended December 31, 2024, the Company recognized a change in fair value resulting in a gain of $ million, respectively, related to the liability-classified warrants prior to meeting the criteria for equity classification.
There were no material liability-classified warrants measured at fair value on a recurring basis using significant unobservable inputs (Level 3) for the year ended December 31, 2025. Changes in the fair value of the liability-classified warrants are recognized as a separate component in the consolidated statement of operations.
F-20
TONIX PHARMACEUTICALS HOLDING CORP.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 7 – OTHER BALANCE SHEET INFORMATION
Components of selected captions in the consolidated balance sheets consist of:
| December
31, 2025 |
December
31, 2024 |
|||||||
| Prepaid expenses and other current assets: | (in thousands) | |||||||
| Contract-related | $ | $ | ||||||
| Prepaid Inventory | ||||||||
| Government grants | ||||||||
| At-the-market receivable | ||||||||
| Non-trade receivables | ||||||||
| Debt interest and fees | ||||||||
| Insurance | ||||||||
| Prescription drug user fee | ||||||||
| Taxes | ||||||||
| Professional fees and other | ||||||||
| $ | $ | |||||||
| Accrued expenses and other current liabilities: | ||||||||
| Contract-related | $ | $ | ||||||
| Compensation and compensation-related | ||||||||
| Gross-to-net deductions | ||||||||
| Professional fees and other | ||||||||
| $ | $ | |||||||
NOTE 8 – DEBT FINANCING
Long-term debt consists of the following:
| December
31, 2025 |
December
31, 2024 |
|||||||
| Term Loan | $ | $ | ||||||
| Less: current portion | ( |
) | ||||||
| Total long-term debt | ||||||||
| Less: unamortized debt discount and deferred financing costs | ( |
) | ||||||
| Total long-term debt, net | $ | $ | ||||||
On
December 8, 2023, the Company entered into a Loan and Guaranty Agreement (the “Loan Agreement”) by and among the Company,
Krele LLC, Tonix Pharmaceuticals, Inc., Jenner and Tonix R&D Center (collectively, the “Loan Parties”), with JGB
Capital, LP, JGB Partners, LP, JGB (Cayman) Port Ellen Ltd., and any other lender from time to time party hereto (collectively,
the “Lenders”), and JGB Collateral LLC, as administrative agent and collateral agent for the Lenders (in such capacity,
“JGB Agent”) for a
Commencing
on
The Loan Agreement provided for voluntary prepayments of the Term Loan, in whole or in part, subject to a prepayment premium. The Term Loan was secured by first priority security interests in the Company’s R&D Center in Frederick, Maryland, the Advanced Development Center in North Dartmouth, Massachusetts, and substantially all of the relevant deposit accounts.
During
the first quarter of 2025, the Company paid $
F-21
TONIX PHARMACEUTICALS HOLDING CORP.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 9 – STOCKHOLDERS’ EQUITY
On
February 5, 2025, the Company effected a
NOTE 10 – REVENUES
Disaggregation of Net Revenues
The Company’s net product revenues are summarized below:
Year ended December 31, |
||||||||
| 2025 | 2024 | |||||||
| Tonmya | $ | $ | ||||||
| Zembrace Symtouch | ||||||||
| Tosymra | ||||||||
| Total product revenues | $ | $ | ||||||
All sales were generated in the United States.
Gross-to-Net Sales Accruals
We record gross-to-net sales accruals for chargebacks, rebates, sales and other discounts, and product returns, which are all customary to the pharmaceutical industry.
Our
provision for gross-to-net allowances was $
NOTE 11 – ASSET PURCHASE AGREEMENT WITH UPSHER-SMITH
On June 30, 2023, the Company completed the acquisition of certain assets from Upsher Smith related to Zembrace SymTouch (sumatriptan injection) 3 mg (“Zembrace”) and Tosymra (sumatriptan nasal spray) 10 mg (“Tosymra”) products.
The
Company has assumed certain obligations of Upsher Smith, including the payment of quarterly royalty payments on annual net sales
from the Business in the U.S. as follows: for Tosymra,
For
Zembrace, royalty payments on annual net sales in the U.S. are
F-22
TONIX PHARMACEUTICALS HOLDING CORP.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
In
addition, the Company has assumed the obligation to pay an additional
NOTE 12 – LICENSE AGREEMENTS WITH UNIVERSITY OF MASSACHUSETTS (UMASS)
On
June 26, 2025, Tonix obtained an exclusive worldwide license from UMass Chan Medical School for the development of TNX-4800 (formerly
known as mAb 2217LS). As of December 31, 2025, other than the upfront fee of $
NOTE 13 – SALE AND PURCHASE OF COMMON STOCK
December 2025 Financing
On December 29, 2025, the Company entered into a securities purchase agreement with an institutional investor, pursuant to which the Company sold shares of common stock and pre-funded warrants to purchase up to shares of common stock. The offering price per share of common stock was $, and the offering price per share of pre-funded warrant was $.
The
offering closed on December 30, 2025. The Company incurred offering expenses of approximately $
2025 Lincoln Park Transaction
On
June 11, 2025, the Company entered into a purchase agreement (the “2025 Purchase Agreement”) and a registration rights
agreement (the “2025 Registration Rights Agreement”) with Lincoln Park. Pursuant to the terms of the 2025 Purchase
Agreement, Lincoln Park has agreed to purchase from the Company up to $
Pursuant
to the terms of the 2025 Purchase Agreement, at the time the Company signed the 2025 Purchase Agreement and the 2025 Registration
Rights Agreement, the Company issued shares of common stock to Lincoln Park as consideration for its commitment to purchase
shares of the Company’s common stock under the 2025 Purchase Agreement. The commitment shares were valued at $
The Company evaluated the 2025 Purchase Agreement under ASC 815-40 Derivatives and Hedging-Contracts on an Entity’s Own Equity as it represents the right to require Lincoln Park to purchase shares of common stock in the future, similar to a put option. The Company concluded that the 2025 Purchase Agreement represents a freestanding derivative instrument that does not qualify for equity classification and therefore requires fair value accounting. The Company analyzed the terms of the contract and concluded that the derivative instrument had insignificant value as of December 31, 2025.
2025 At-the-Market Offerings
On
June 11, 2025, the Company entered into a Sales Agreement (the “2025 Sales Agreement”), with A.G.P./Alliance Global
Partners (“AGP”) pursuant to which the Company may issue and sell, from time to time, shares of common stock having
an aggregate offering price of up to $
2024 At-the-Market Offerings
On
July 30, 2024, the Company entered into a Sales Agreement (the “2024 Sales Agreement”), with AGP pursuant to which the Company
may issue and sell, from time to time, shares of common stock having an aggregate offering price of up to $
F-23
TONIX PHARMACEUTICALS HOLDING CORP.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
July 2024 Financing
On July 9, 2024, the Company entered into a securities purchase agreement with certain institutional and retail investors, pursuant to which the Company sold shares of common stock and pre-funded warrants to purchase up to shares of common stock. The offering price per share of common stock was $, and the offering price per share of pre-funded warrant was $.
The
offering closed on July 10, 2024. The Company incurred offering expenses of approximately $
June 2024 Financings
On June 12, 2024, the Company entered into a securities purchase agreement with certain investors, pursuant to which the Company sold shares of common stock and pre-funded warrants to purchase up to shares of common stock. The offering price per share of common stock was $, and the offering price per share of pre-funded warrant was $.
The
offering closed on June 13, 2024. The Company incurred offering expenses of approximately $
On June 27, 2024, the Company entered into a securities purchase agreement with certain institutional and retail investors, pursuant to which the Company sold shares of common stock and pre-funded warrants to purchase up to shares of common stock. The offering price per share of common stock was $, and the offering price per share of pre-funded warrant was $.
The
offering closed on June 28, 2024. The Company incurred offering expenses of approximately $
March 2024 Financing
On
March 28, 2024, the Company entered into an agreement to sell shares of common stock, pre-funded warrants to purchase up
to shares of common stock, and accompanying Series E warrants to purchase up to shares of common stock with an exercise
price of $
The
Company incurred expenses of approximately $
Additionally,
with the closing of the financing on April 1, 2024, the Company entered into warrant amendments (collectively, the “Warrant
Amendments”) with certain holders of its common warrants (referred to herein as the “Existing Warrants”). The
Company agreed to amend the exercise price of each Existing Warrant to $
F-24
TONIX PHARMACEUTICALS HOLDING CORP.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The
Company evaluated the Warrant Amendments as of April 1, 2024, and determined that the potential adjustment to the exercise price
that is contingent on stockholder approval precluded the Existing Warrants from being indexed to the Company’s own stock,
and as a result, did not meet the criteria for equity classification under ASC 815-40. The Company accounted for the incremental
fair value of the Warrant Amendments of $
The liability-classified Series D Warrants and all of the Series C Warrants were presented within non-current liabilities on the consolidated balance sheets as of December 31, 2023, and were adjusted to fair value through January 25, 2024, when the warrants were reclassified to equity. Changes in the fair value of the liability-classified warrants were recognized as a separate component in the consolidated statement of operations.
Stock repurchases
In September 2024, the Board of Directors approved a 2024 share repurchase program pursuant to which the Company may repurchase up to $.0 million in value of its outstanding common stock from time to time on the open market and in privately negotiated transactions subject to market conditions, share price and other factors. In November 2025, the amount increased to $.0 million.
During the year ended December 31, 2025, the Company repurchased shares of its common stock outstanding under the 2024 share repurchase at prices ranging from $ to $ per share for a gross aggregate cost of approximately $ million. The repurchased shares were immediately retired.
| Year
Ended December 31, 2025 |
Year
Ended December 31, 2024 |
|||||||
| Total cost of repurchased shares (in thousands) | $ | $ | — | |||||
| Shares repurchased | — | |||||||
| Weighted average price per share | $ | $ | — | |||||
The timing and amount of any shares repurchased will be determined based on the Company’s evaluation of market conditions and other factors and the New Share Repurchase Program may be discontinued or suspended at any time. Repurchases will be made in accordance with the rules and regulations promulgated by the Securities and Exchange Commission and certain other legal requirements to which the Company may be subject. Repurchases may be made, in part, under a Rule 10b5-1 plan, which allows stock repurchases when the Company might otherwise be precluded from doing so.
On May 1, 2020, the Company’s stockholders approved the Tonix Pharmaceuticals Holding Corp. Amended and Restated 2020 Stock Incentive Plan (“Amended and Restated 2020 Plan”).
Under the terms of the Amended and Restated 2020 Plan, the Company may issue (1) stock options (incentive and nonstatutory), (2) restricted stock, (3) stock appreciation rights (“SARs”), (4) RSUs, (5) other stock-based awards, and (6) cash-based awards. The Amended and Restated 2020 Plan initially provided for the issuance of up to shares of common stock, which amount will be increased to the extent that awards granted under the Plans are forfeited, expire or are settled for cash (except as otherwise provided in the Amended and Restated 2020 Plan). In addition, the Amended and Restated 2020 Plan contains an “evergreen provision” providing for an annual increase in the number of shares of our common stock available for issuance under the Amended and Restated 2020 Plan on January 1 of each year for a period of ten years, commencing on January 1, 2021 and ending on (and including) January 1, 2030, in an amount equal to the difference between (x) twenty percent (%) of the total number of shares of common stock outstanding on December 31st of the preceding calendar year, and (y) the total number of shares of common stock reserved under the Amended and Restated 2020 Plan on December 31st of such preceding calendar year (including shares subject to outstanding awards, issued pursuant to awards or available for future awards). On May 8, 2025, the Company’s stockholders approved the addition of shares to the Company’s Amended and Restated 2020 Plan.
F-25
TONIX PHARMACEUTICALS HOLDING CORP.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The Board of Directors determines the exercise price, vesting and expiration period of the grants under the Amended and Restated 2020 Plan. However, the exercise price of an incentive stock option may not be less than % of fair value of the common stock at the date of the grant for a 10% or more shareholder and % of fair value for a grantee who is not a 10% shareholder. The fair value of the common stock is determined based on quoted market price or in absence of such quoted market price, by the Board of Directors in good faith. Additionally, the expiration period of grants under the Amended and Restated 2020 Plan may not be more than . As of December 31, 2025, there were options available for future grants under the Amended and Restated 2020 Plan.
General
| Shares | Weighted-
Average Exercise Price |
Weighted-Average Remaining Contractual Term | Aggregate Intrinsic Value | |||||||||||||
| Outstanding at December 31, 2023 | $ | $ | — | |||||||||||||
| Grants | $ | — | — | |||||||||||||
| Exercised | — | — | ||||||||||||||
| Forfeitures or expirations | ( |
) | ||||||||||||||
| Outstanding at December 31, 2024 | $ | $ | — | |||||||||||||
| Grants | $ | — | $ | |||||||||||||
| Exercised | — | — | ||||||||||||||
| Forfeitures or expirations | ( |
) | $ | |||||||||||||
| Outstanding at December 31, 2025 | $ | $ | — | |||||||||||||
| Exercisable at December 31, 2025 | $ | $ | — | |||||||||||||
The aggregate intrinsic value in the preceding table represents the total pretax intrinsic value, based on options with an exercise price less than the Company’s closing stock price at the respective dates.
The weighted average fair value of options granted during the year ended December 31, 2025, and December 31, 2024 was $ and $ per share, respectively.
The Company measures the fair value of stock options on the date of grant, based on the Black Scholes option pricing model using certain assumptions discussed below, and the closing market price of the Company’s common stock on the date of the grant. The fair value of the award is measured on the grant date. of most stock options granted pursuant to the Plans vest 12 months from the date of grant and each month thereafter for 24 months and expire from the date of grant. In addition, the Company issues options to directors which vest over a period. The Company also issues premium options to executive officers which have an exercise price greater than the grant date fair value and has issued performance-based options which vest when target parameters are met or probable of being met, subject in each case to a minimum service period prior to vesting. Stock-based compensation expense related to awards is amortized over the applicable service period using the straight-line method.
| Year
Ended December 31, 2025 |
Year
Ended December 31, 2024 |
|||||||
| Risk-free interest rate | % to | % | % to | % | ||||
| Expected term of option | to years | to years | ||||||
| Expected stock price volatility | % to | % | % - | % | ||||
| Expected dividend yield | ||||||||
F-26
TONIX PHARMACEUTICALS HOLDING CORP.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The risk-free interest rate is based on the yield of Daily U.S. Treasury Yield Curve Rates with terms equal to the expected term of the options as of the grant date. The expected term of options is determined using the simplified method, as provided in an SEC Staff Accounting Bulletin, and the expected stock price volatility is based on the Company’ historical stock price volatility.
Stock-based compensation expense relating to options granted of $ million, of which $ million and $ million, related to General and Administration and Research and Development, respectively was recognized for the year ended December 31, 2025. Stock-based compensation expense relating to options granted of $ million, of which $ million and $ million, related to General and Administration and Research and Development, respectively was recognized for the year ended December 31, 2024.
As of December 31, 2025, the Company had approximately $ million of total unrecognized compensation cost related to non-vested awards granted under the Plans, which the Company expects to recognize over a weighted average period of years.
Employee Stock Purchase Plans
On May 5, 2023, the Company’s stockholders approved the Tonix Pharmaceuticals Holdings Corp. 2023 Employee Stock Purchase Plan. (the “2023 ESPP”), which was replaced by the Tonix Pharmaceuticals Holdings Corp. 2025 Employee Stock Purchase Plan (the “2025 ESPP”, and together with the 2023 ESPP, the “ESPP Plans”), which was approved by the Company’s stockholders on May 8, 2025.
The 2025 ESPP allows eligible employees to purchase up to an aggregate of shares of the Company’s common stock. Under the 2025 ESPP, on the first day of each offering period, each eligible employee for that offering period has the option to enroll for that offering period, which allows the eligible employees to purchase shares of the Company’s common stock at the end of the offering period. Each offering period under the 2025 ESPP is for six months, which can be modified from time to time. Subject to limitations, each participant will be permitted to purchase a number of shares determined by dividing the employee’s accumulated payroll deductions for the offering period by the applicable purchase price, which is equal to percent of the fair market value of our common stock at the beginning or end of each offering period, whichever is less. A participant must designate in his or her enrollment package the percentage (if any) of compensation to be deducted during that offering period for the purchase of stock under the 2025 ESPP, subject to the statutory limit under the Code.
The 2023 ESPP allows eligible employees to purchase up to an aggregate of shares of the Company’s common stock. Under the 2023 ESPP, on the first day of each offering period, each employee eligible for that offering period has the option to enroll for that offering period, which allows the eligible employees to purchase shares of the Company’s common stock at the end of the offering period. Each offering period under the 2023 ESPP is for six months, which can be modified from time-to-time. Subject to limitations, each participant will be permitted to purchase a number of shares determined by dividing the employee’s accumulated payroll deductions for the offering period by the applicable purchase price, which is equal to percent of the fair market value of our common stock at the beginning or end of each offering period, whichever is less. A participant must designate in his or her enrollment package the percentage (if any) of compensation to be deducted during that offering period for the purchase of stock under the 2023 ESPP, subject to the statutory limit under the Code. As of December 31, 2025, shares were available for future sales under the 2023 ESPP and shares were available under the 2025 ESPP.
The ESPP Plans are considered compensatory plans with the related compensation cost expensed over the six-month offering period. For the year ended December 31, 2025, and 2024, $ and $, respectively, was expensed. In January 2024, shares that were purchased as of December 31, 2023, under the 2022 ESPP, were issued. As of June 30, 2024, approximately $ of employee payroll deductions had accumulated and had been recorded in accrued expenses. In July 2024, shares that were purchased as of June 30, 2024, under the 2022 ESPP, were issued. As of December 31, 2025, approximately $ of employee payroll deductions had accumulated and had been recorded in accrued expenses. In January 2026, shares that were purchased as of December 31, 2025, under the 2022 ESPP, were issued.
NOTE 15 – STOCK WARRANTS
The following table summarizes information with respect to outstanding warrants to purchase common stock of the Company at December 31, 2025:
| Exercise | Number | Expiration | |||||||
| Price | Outstanding | Date | |||||||
| $ | |||||||||
| $ | |||||||||
| $ | |||||||||
| $ | |||||||||
| $ | |||||||||
| $ | |||||||||
| $ | |||||||||
| $ | |||||||||
F-27
TONIX PHARMACEUTICALS HOLDING CORP.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
During
the year ended December 31, 2025, ; (as the Company received FDA acceptance of our NDA filing), and warrants with
an exercise price of $
During the year ended December 31, 2025, warrants were exercised.
During the year ended December 31, 2024, prefunded common warrants were exercised.
For
the year ended December 31, 2024, and warrants with an exercise price of $
For
the year ended December 31, 2024, and warrants with an exercise price of $
Additionally,
with the closing of the financing on April 1, 2024, the Company entered into the Warrant Amendments (as defined in Note 13) with
certain holders of its warrants to purchase common stock, agreeing to amend the exercise price of each Existing Warrant to $
NOTE 16 – LEASES
The Company has various operating lease agreements, which are primarily for office space. These agreements frequently include one or more renewal options and require the Company to pay for utilities, taxes, insurance and maintenance expense. No lease agreement imposes a restriction on the Company’s ability to engage in financing transactions or enter into further lease agreements. At December 31, 2025, the Company has right-of-use assets of $ million and a total lease liability for operating leases of $ million of which $ million is included in long-term lease liabilities and $ million is included in current lease liabilities.
At December 31, 2025, future minimum lease payments for operating leases with non-cancelable terms of more than one year were as follows (in thousands):
| Year Ending December 31, | |||||
| 2026 | $ | ||||
| 2027 | |||||
| 2028 | |||||
| 2029 | |||||
| 2030 and thereafter | |||||
| Included interest | ( |
) | |||
| $ | |||||
During
the year ended December 31, 2025, the Company entered into a new operating lease and lease amendments. The lease commencement
during the fourth quarter of 2025, at which time the Company recognized an additional operating lease liability of approximately
$
F-28
TONIX PHARMACEUTICALS HOLDING CORP.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Other information related to leases is as follows:
Operating
lease expense was $
Other information related to leases is as follows:
| Cash paid for amounts included in the measurement of lease liabilities: | Year
Ended December 31, 2025 |
Year
Ended December 31, 2024 |
||||||
| Operating cash flow from operating leases (in thousands) | $ | $ | ||||||
| Weighted Average Remaining Lease Term | ||||||||
| Operating leases | ||||||||
| Weighted Average Discount Rate | ||||||||
| Operating leases | % | % | ||||||
NOTE 17 – COMMITMENTS
Contractual agreements
The
Company has entered into contracts with various contract research organizations with outstanding commitments aggregating approximately
$
The
Company has entered into various exclusive license agreements with various institutions with the right to sublicense, certain
patents, technical information and material, and to develop and commercialize products thereunder. In addition to any upfront
payments already paid, the Company may be obligated to pay milestone fees ranging from $
Defined contribution plan
The
Company has a qualified defined contribution plan (the “401(k) Plan”) pursuant to Section 401(k) of the Code, whereby
all eligible employees may participate. Participants may elect to defer a percentage of their annual pretax compensation to the
401(k) Plan, subject to defined limitations. The Company is required to make contributions to the 401(k) Plan equal to
NOTE 18 – INCOME TAXES
Components of the net loss consist of the following (in thousands):
| Year ended December 31, | ||||||||
| 2025 | 2024 | |||||||
| Foreign | $ | ( |
) | $ | ( |
) | ||
| Domestic | ( |
) | ( |
) | ||||
| Total | $ | ( |
) | $ | ( |
) | ||
In 2025, the foreign losses are primarily comprised
of $
The
Company adopted ASU 2023-09 and applied the new disclosure requirements prospectively for the year ended December 31, 2025. A reconciliation
of the anticipated income tax benefit computed by applying the statutory federal income tax rate of
| Year Ended December 31, | ||||||||
| 2025 | ||||||||
| Amount | Percent | |||||||
| Tax at U.S. Statutory Rate | ( | ) | % | |||||
| State and Local Income Taxes, Net of Federal Income Tax Effect | % | |||||||
| Foreign Tax Effects | ||||||||
| Ireland | ||||||||
| Change in Foreign Valuation Allowance | - | % | ||||||
| Statutory tax rate difference between Ireland and United States | - | % | ||||||
| Other Foreign Jurisdiction | ( | ) | % | |||||
| Change in Domestic Valuation Allowance | - | % | ||||||
| Nontaxable and Nondeductible items | % | |||||||
| Attributable reduction from control change | - | % | ||||||
| Others | - | % | ||||||
| Effective Tax Rate | % | |||||||
A reconciliation of the effect of applying the federal statutory rate to the net loss and the effective income tax rate used to calculate the Company’s income tax provision is as follows:
| Year Ended December 31, | ||||||||
| 2024 | 2023 | |||||||
| Statutory federal income tax | ( |
)% | ( |
)% | ||||
| Change in valuation allowance | % | % | ||||||
| Permanent differences | ( |
)% | % | |||||
| Foreign loss not subject to income tax | % | % | ||||||
| Attribute reduction from control change | % | % | ||||||
| Other | % | % | ||||||
| Income Tax Provision | % | % | ||||||
Deferred tax assets (liabilities) and related valuation allowance as of December 31, 2025 and 2024 were as follows (in thousands):
| December 31, | ||||||||
| 2025 | 2024 | |||||||
| Deferred tax assets/(liabilities): | ||||||||
| Net operating loss carryforward | $ | $ | ||||||
| Stock-based compensation | ||||||||
| Fixed assets | ||||||||
| Other | ||||||||
| Total deferred assets | ||||||||
| Valuation allowance | ( |
) | ( |
) | ||||
| Net deferred tax assets | $ | $ | ||||||
The Company has incurred
research and development (“R&D”) expenses, a portion of which qualifies for tax credits. The Company conducted an R&D credit
study to quantify the amount of credits and has claimed an R&D credit on its 2014-2017 tax returns. A portion of these R&D credit
carryforwards are subject to annual limitations in their use in accordance with Internal Revenue Service Code ("IRC") section
383. The R&D credit carryforwards at December 31, 2024 were reduced to $
In 2025, the Company completed an analysis under Section 382 of the Internal Revenue Code with respect to its net operating loss carryforwards. Based on this analysis, the Company determined that all historical loss carryforwards generated prior to December 31, 2024 are subject to limitation and effectively unavailable for use under Section 382. Net operating losses generated after December 31, 2024 are not subject to the Section 382 limitation.
As of December 31, 2025,
the Company had net operating loss carryforwards consisting of $
Management assesses the
available positive and negative evidence to estimate if sufficient future taxable income will be generated to use the existing deferred
tax assets. A significant piece of objective negative evidence evaluated was the cumulative loss incurred over the three-year period ended
December 31, 2025. Such objective evidence limits the ability to consider other subjective evidence such as our projections for future
growth. As such, the Company has determined that it is not more likely than not that the deferred tax assets will be realized and accordingly,
has provided a full valuation allowance against its gross deferred tax assets. The increase/(decrease) in the valuation allowance for
the year ended December 31, 2025 was an increase of $
The Company recognizes interest accrued related to unrecognized tax benefits and penalties as income tax expense. However, as of December 31, 2025 there are no unrecognized tax benefits recorded. The Company is subject to taxation in the United States and various states and foreign jurisdictions. As of December 31, 2025, the Company's tax returns remain open and subject to examination by the tax authorities for the tax years 2022 and after.
The Company has not made payments or received refunds for income taxes for the years ended December 31, 2025 and 2024.
On July 4, 2025 the OBBBA was enacted into law. The legislation made several changes to the U.S. tax code, including the return of 100% bonus depreciation, the ability to immediately deduct domestic research and development costs, a more favorable rule for deducting interest expenses, and updated to international tax rules around global intangible low-taxed income and foreign-derived intangible income. The Company has evaluated the impact of the new tax provision and determined it to have an immaterial impact.
NOTE 19 – SUBSEQUENT EVENTS
On February 24, 2026, the Company granted options to purchase an aggregate of shares of the Company’s common stock to employees with an exercise price of . Additionally, the Company granted options to purchase shares of the Company’s common stock to certain employees with an exercise price of .
Subsequent
to December 31, 2025, the Company has sold million shares of common stock under the Sales Agreement, for net proceeds of approximately
$
F-29
ITEM 9 – CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURES
None.
ITEM 9A – CONTROLS AND PROCEDURES
Management’s evaluation of disclosure controls and procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures pursuant to Rule 13a-15 under the Exchange Act. In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives. In addition, the design of disclosure controls and procedures must reflect the fact that there are resource constraints, and that management is required to apply its judgment in evaluating the benefits of possible controls and procedures relative to their costs.
Based on management’s evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures are designed at a reasonable assurance level and are effective to provide reasonable assurance that information we are required to disclose in reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in SEC rules and forms, and that such information is accumulated and communicated to our management, including our chief executive officer and chief financial officer, as appropriate, to allow timely decisions regarding required disclosure.
Management’s report on internal control over financial reporting.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting for our company. Internal control over financial reporting is defined in Rule 13a-15(f) and 15d-15(f) promulgated under the Exchange Act, as a process designed by, or under the supervision of, a company’s principal executive and principal financial officer and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
| (1) | pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; | |
| (2) | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made in accordance with authorizations of management and directors of the company; and | |
| (3) | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’s assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible enhancements to controls and procedures.
We conducted an evaluation of the effectiveness of internal control over financial reporting based on the framework in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on this evaluation, our principal executive officer and principal financial officer conclude that, at December 31, 2025, our internal control over financial reporting was effective.
This Annual Report does not include an attestation report by EisnerAmper LLP, our independent registered public accounting firm regarding internal control over financial reporting. As a smaller reporting company, our management’s report was not subject to attestation by our registered public accounting firm pursuant to rules of the Securities and Exchange Commission that permit us to provide only management’s report in this Annual Report.
99
Changes in internal control over financial reporting.
There were no changes in our internal control over financial reporting that occurred during the year ended December 31, 2025 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
ITEM 9B – OTHER INFORMATION
During
the fiscal quarter ended December 31, 2025, our officers or directors, as those terms are defined in Rule 16a-1(f),
ITEM 9C – DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not Applicable.
100
PART III
ITEM 10 – DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The Board of Directors elects our executive officers annually. A majority vote of the directors who are in office is required to fill vacancies. Each director shall be elected for the term of one year and until his successor is elected and qualified or until his earlier resignation or removal. Our directors and executive officers are as follows:
| NAME | AGE | CURRENT POSITION | ||
| Seth Lederman | 68 | President, CEO and Chairman of the Board of Directors | ||
| Richard Bagger | 65 | Director | ||
| Margaret Smith Bell | 66 | Director | ||
| David Grange | 78 | Director | ||
| James Hunter | 70 | Director | ||
| Adeoye Olukotun | 81 | Director | ||
| Newcomb Stillwell | 69 | Director | ||
| Carolyn Taylor | 66 | Director | ||
| James Treco | 70 | Lead Director | ||
| Jessica Morris | 48 | Chief Operating Officer | ||
| Bradley Saenger | 52 | Chief Financial Officer and Treasurer | ||
| Gregory Sullivan | 60 | Chief Medical Officer and Secretary | ||
| Siobhan Fogarty | 57 | Chief Technical Officer |
The following information with respect to the principal occupation or employment of each nominee for director, the principal business of the corporation or other organization in which such occupation or employment is carried on, and such nominee’s business experience during the past five years, as well as the specific experiences, qualifications, attributes and skills that have led the Board to determine that such Board members should serve on our Board, has been furnished to the Company by the respective director nominees:
Seth Lederman, MD became our President, Chief Executive Officer, Chairman of the Board and a Director in October 2011. Dr. Lederman founded Tonix Pharmaceuticals, Inc., a wholly-owned subsidiary, in 2007 and has acted as its Chairman of the Board of Directors since its inception, and as President since 2010. Prior to becoming a biopharma entrepreneur, Dr. Lederman served as an Associate Professor at Columbia University, where he directed basic science research in molecular immunology, infectious diseases and the development of therapeutics for autoimmune diseases. In addition to his research, Dr. Lederman served as attending physician in the Edward Daniels Arthritis and Autoimmunity Clinic on the Medical Service at Columbia Presbyterian Hospital. Dr. Lederman received his BA degree in Chemistry from Princeton University in 1979 and his M.D. from Columbia University in 1983. Dr. Lederman’s significant experience with drug discovery, development and commercialization and his experience as an biotechnology executive director were instrumental in his selection as a member of the Board.
101
Richard Bagger became a Director in June 2020. Mr. Bagger has been a Partner and Executive Director of Christie 55 Solutions, LLC, a New Jersey based consulting firm, since January 2020. Mr. Bagger has also been an Adjunct Faculty member at Rutgers University since 2018. From 2012 through 2019, Mr. Bagger was Executive Vice President of Corporate Affairs and Market Access for Celgene Corporation (NASDAQ: CELG), a global biopharmaceutical company, as well as a member of its Executive Committee. From 1993 to 2010, Mr. Bagger held roles of increasing responsibility with Pfizer Inc. (NYSE: PFE), a global pharmaceutical company, and served as Senior Vice President, Worldwide Public Affairs and Policy, from 2006 to 2009. Prior to joining Pfizer, Mr. Bagger was Assistant General Counsel of Blue Cross and Blue Shield of New Jersey, a health insurer, and practiced law with the law firm of McCarter & English. Mr. Bagger served as Board Chair of the National Pharmaceutical Council for 2019 and is a member of the Board of Directors of the U.S. Chamber of Commerce. He is also on the advisory board for the Lerner Center for the Study of Pharmaceutical Management Issues at Rutgers University Business School. Mr. Bagger received an A.B. degree from Princeton University’s School of Public and International Affairs and a J.D. degree from Rutgers University Law School. Mr. Bagger’s extensive healthcare and public policy experience were instrumental in his selection as a member of the Board.
Margaret Smith Bell became a Director in September 2017. Previously, Ms. Bell was a Vice President at Standard Life Investments where she was a portfolio manager and health care equity analyst. Ms. Bell was also a Managing Director at Putnam Investments and served as a senior health care analyst and a portfolio manager of the Putnam Health Sciences Trust. Ms. Bell was an analyst and vice president at State Street Research and a research analyst at Alex. Brown & Sons, Inc. Ms. Bell is a past member of the Board of Overseers at Beth Israel Deaconess Medical Center. Ms. Bell holds a B.A. from Wesleyan University and an M.B.A. from the Wharton School at the University of Pennsylvania. Ms. Bell’s extensive healthcare and investment banking experience were instrumental in her selection as a member of the Board.
Major General David Grange (U.S. Army, retired) became a director in February 2018. MG Grange has been President and founder of Osprey Global Solutions, LLC (“OGS”), a Service Disabled Veterans Organization, since 2011. MG Grange was Chief Executive Officer of Pharm-Olam International, Ltd., a contract research organization, from April 2017 to October 2019. Prior to founding OGS, MG Grange was a member of the Board of Pharmaceutical Product Development, Inc. (Nasdaq: PPDI), a contract research organization, from 2003 to 2009, and Chief Executive Officer from 2009 to 2011.
Prior to PPDI MG Grange served in the McCormick Tribune Foundation for 10 years, most recently as Chief Executive Officer and President, where he also oversaw the support of Veteran Programs. MG Grange served 30 years in the U.S. Army as a Ranger, Green Beret, Aviator, Infantryman and a member of special operating units. At the Pentagon, he was Director of Army Current Operations, Readiness, and Mobilization. MG Grange commanded the Ranger Regiment and the First Infantry Division (the Big Red One). MG Grange holds a master’s degree in Public Service from Western Kentucky University. MG Grange’s extensive experience in the pharmaceutical industry and service with the U.S. military was instrumental in his selection as a member of our Board.
James Hunter became a Director in June 2025. Mr. Hunter served as executive Vice President of Commercial Operations at Tonix Pharmaceuticals and President of Tonix Medicines from June 2023 to December 2024. Mr. Hunter was CEO and Co-founder of Validus Pharmaceuticals from 2007 to 2018. Before co-founding Validus, Mr. Hunter was Executive Director of Neuroscience Sales at Novartis Pharmaceuticals. Previously, Mr. Hunter served as Executive Director of the Northeast Business Unit at Ciba Geigy Pharmaceuticals. Mr. Hunter received his B.S. from Seton Hall University and earned his M.B.A from Fairleigh Dickinson University. Mr. Hunter’s extensive experience in the pharmaceutical industry was instrumental in his selection as a member of our Board.
Adeoye Olukotun, MD became a Director in September 2018. Dr. Olukotun is a board member of Arrowhead Pharmaceuticals (ARWR), a publicly traded biopharmaceutical company. Dr. Olukotun has been the Chief Executive Officer of CR Strategies, LLC, a medical products consulting company, since 2000. Dr. Olukotun was the Chief Executive Officer of Genesis Unicorn Corporation, a special acquisition company listed on Nasdaq (GENQU) that became Genesis Unicorn Capital Corp. (GENQ), and later became ESGL Holdings Ltd trading on Nasdaq (ESGL). Dr. Olukoton was the Chief Executive Officer of EpiGen Pharmaceuticals, Inc., a pharmaceutical company, from 2014 to January of 2018. Dr. Olukotun served as Vice Chairman of CardoVax, Inc., a pharmaceutical company, from 2012 to 2016, and as its Chief Executive Officer from 2006 to 2012. He is also co-founder of VIA Pharmaceuticals, Inc., a pharmaceutical company, and served as the company’s Chief Medical Officer from 2004 to 2008. Dr. Olukotun is a member of the board of directors of Arrowhead Pharmaceuticals. Dr. Olukotun received his B.A. from University of North Carolina, Chapel Hill, his M.D. from Albert Einstein College of Medicine, and his M. P.H. from Harvard School of Public Health. Dr. Olukotun’s extensive medical background and experience in the pharmaceutical industry was instrumental in his selection as a member of our Board.
R. Newcomb Stillwell became a director in March 2023. Mr. Stillwell has held positions of varying responsibility at the law firm of Ropes & Gray LLP from 1984 to 2021, including, most recently, as co-managing partner of the Ropes & Gray Boston office. Mr. Stillwell graduated from Harvard Law School and earned an A.B. from Princeton University. Mr. Stillwell’s extensive advisory experience on numerous transactions in the life science and healthcare sectors was instrumental in his selection as a member of the Board.
102
Carolyn Taylor became a director in July 2021. Ms. Taylor was general counsel of Strike Protocols Inc., a financial technology company, from 2019 to 2020, and held positions of varying responsibility, including partner, and most recently, of counsel, at the law firm of Covington & Burling LLP from 1989 to 2000 and 2004 to 2015. From 2000 to 2003, Ms. Taylor served as Executive Vice President and General Counsel of Longitude, Inc., a financial services company. Ms. Taylor graduated from Columbia Law School and earned a B.A. from Brown University. Ms. Taylor’s broad transactional experience was instrumental in her selection as a member of the Board.
James Treco became a director in February 2019 and has been our Lead Director since March 2020. Mr. Treco continues to be involved with several small clinical research companies operating out of the Hanover, New Hampshire area. Mr. Treco has been a Managing Partner at First Chicago Advisors, Inc., a boutique financial advisory firm where he advised executives and boards of directors of a wide range of companies, from global, large-cap companies to emerging companies, from 2009 to 2012 and from 2014 to 2024. From 2012 to 2013 Mr. Treco was an investment banker with Gleacher & Company, a company that previously operated an investment banking business, providing corporate and institutional clients with strategic and financial advisory services. Mr. Treco held various positions of increasing responsibility at Salomon Brothers/Citigroup from 1984 to 2008, where he used his extensive experience in the global capital markets to advise a wide range of clients. Mr. Treco holds a B.A. from Yale University and an M.B.A. from the Stanford University Graduate School of Business. Mr. Treco’s extensive healthcare and investment banking experience were instrumental in his selection as a member of the Board.
Jessica Morris is our Chief Operating Officer and has worked for the Company since April 2013, first as a consultant (April 2013 – September 2013), then as SVP of Finance (September 2013 – October 2015), followed by Chief Administrative Officer (October 2015 – January 2016), Acting Chief Financial Officer (January 2016 – February 2016), and Executive Vice President, Operations (February 2016 – January 2018). Prior to joining the Company, Ms. Morris was a Vice President in investment management at Zhong Rong Group. Previously, Ms. Morris was a Senior Associate in the Sponsor Finance Group at American Capital, a Vice President of the mezzanine debt fund at Calvert Street Capital Partners, an Associate in the commercial finance department of Silicon Valley Bank, and a Financial Analyst in the investment banking group at Deutsche Bank. Ms. Morris earned a B.S. in Commerce and a B.A. in Music from the University of Virginia, where she was an Echols Scholar.
Bradley Saenger, CPA became Chief Financial Officer in February 2016 and joined Tonix in May 2014, where he held positions of increasing responsibility. Since November 2015, Mr. Saenger has been a director of Tonix Pharma Holdings Limited. Between June 2013 and March 2014, Mr. Saenger worked for Shire Pharmaceuticals as a consultant in the financial analyst research and development group. Between February 2013 and May 2013, Mr. Saenger worked for Stewart Health Care System, formerly a private, for-profit hospital operator, as a financial consultant. Between October 2011 and December 2012, Mr. Saenger was an Associate Director of Accounting at Vertex Pharmaceuticals, Inc., a publicly traded biotech company (Nasdaq: VRTX). Between January 2005 and September 2011, Mr. Saenger worked for Alere Inc., formerly a global manufacturer of rapid point-of-care diagnostic tests that was acquired by Abbott Laboratories, as a Manager of Corporate Accounting and Consolidations (2007 – 2011) and Manager of Financial Reporting (2005 – 2006). Mr. Saenger also worked for PricewaterhouseCoopers LLP, Shifren Hirsowitz, public accountants and auditors in Johannesburg, South Africa, Investec Bank in Johannesburg, South Africa and Norman Sifris and Company, public accountants and auditors in Johannesburg, South Africa. Mr. Saenger received his Bachelor’s and Honors’ degrees in Accounting Science from the University of South Africa. Mr. Saenger is a Chartered Accountant in South Africa and a Certified Public Accountant in the Commonwealth of Massachusetts.
Gregory Sullivan, MD became our Chief Medical Officer on June 3, 2014 and our Secretary in March 2017. Prior to becoming our Chief Medical Officer, he served on our Scientific Advisory Board since October 2010, and had also provided ad hoc consulting services. Previously, Dr. Sullivan had been a member of the faculty of Columbia University since July 1999, where he served as an Assistant Professor of Psychiatry in the Department of Psychiatry at Columbia University Medical Center (“CUMC”) until June 2014. Between June 1997 and August 2014, Dr. Sullivan maintained a part-time psychiatry practice. He served as a Research Scientist at the New York State Psychiatric Institute (“NYSPI”) from December 2006 to June 2014. He Dr. Sullivan also served as a member of the Institutional Review Board of the NYSPI from January 2009 to June 2014. As Principal Investigator and Co-Investigator on several human studies of PTSD, Dr. Sullivan has administered the recruitment, biological assessments, treatment, and safety of participants with PTSD in clinical trials of the disorder. He has published more than 50 articles and chapters on research topics ranging from stress and anxiety disorders to abnormal serotonin receptor expression in depression, PTSD and panic disorder. He is a recipient of grants from the National Institute of Mental Health ("NIMH"), the Anxiety Disorders Association of America, NARSAD, the Dana Foundation, and the American Foundation for Suicide Prevention. Dr. Sullivan received a BA in Biology from the University of California, Berkeley, and received his MD from the College of Physicians & Surgeons at Columbia University. He completed his residency training in psychiatry at CUMC, and then a two-year NIMH-sponsored research fellowship in anxiety and affective disorders before joining the faculty at Columbia.
Siobhan Fogarty became our Chief Technical Officer in February 2025, prior to which. Siobhan has worked for she held roles of increasing responsibility Tonix Pharma Limited, a wholly-owned subsidiary of the Company, since September 2016, holding roles with increasing responsibility, most recently as Executive Vice President, Product Development, since February 2021, and prior to that as Vice President, Product Development, since February 2019. Ms. Fogarty started her career with Elan Corporation, an Irish pharmaceutical company, as a formulation scientist. Ms. Fogarty moved to Glaxo SmithKline, a publicly traded pharmaceutical company (NYSE: GSK), as a manufacturing strategist post following the merger of Glaxo and SmithKline Beecham. Ms. Fogarty established European product development sites for Fuisz Technologies, a privately held technology company, and Biovail Corporation, a privately held specialty pharmaceutical company. Subsequently Ms. Fogarty started her own consultancy company, eMSc, continuing to consult with pharmaceutical companies in product development and implementation of a phased approach to quality. Ms. Fogarty earned a Bachelor of Science in Industrial Chemistry from the University of Limerick, and a Masters in Pharmaceutical Science from the School of Pharmacy, Trinity College Dublin.
103
Directors serve until the next annual meeting of shareholders or until their successors are elected and qualified. Officers serve at the discretion of the Board.
Board Independence
The Board has determined that (i) because Seth Lederman is an executive officer of the Company, he has a relationship which, in the opinion of the Board, would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and is not an “independent director” as defined in the Marketplace Rules of The NASDAQ Stock Market, (ii) because James Hunter was a former employee of the Company, he has a relationship which, in the opinion of the Board, would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and is not an “independent director” as defined in the Marketplace Rules of The NASDAQ Stock Market, and (iii) Richard Bagger, Margaret Smith Bell, David Grange, Adeoye Olukotun, Newcomb Stillwell, Carolyn Taylor and James Treco are each an independent director as defined in the Marketplace Rules of The NASDAQ Stock Market.
Board Leadership Structure
Our CEO also serves as the chairman of the Board. An independent director serves as the Board’s lead director. This structure allows one person to speak for and lead both the Company and the Board, while also providing for effective independent board oversight through an independent lead director. Having Dr. Lederman, our CEO, serve as Chairman creates clear and unambiguous authority, which is essential to effective management. Our Board and management can respond more effectively to a clearer line of authority. By designating our CEO as its Chairman, our Board also sends as an important signal to our employees and shareholders about who is accountable. Further, since Dr. Lederman is the founder of our Company and is an inventor on key patents and patent applications underlying our programs, we believe that Dr. Lederman is best-positioned to set our Board’s agenda and provide leadership.
We have established the position of lead director, which has been held by Mr. Treco since 2020. The lead director has the following responsibilities, as detailed in the Lead Director charter, adopted by the Board (and also performs any other functions the Board may request):
| ● | Board leadership — provides leadership to the Board in any situation where the chairman’s role may be, or may be perceived to be, in conflict, and also chairs meetings when the chairman is absent; |
| ● | Leadership of independent director meetings — leads independent director meetings, which take place without any management directors or Tonix employees present; |
| ● | Additional meetings — calls additional independent director meetings as needed; |
| ● | Chairman-independent director liaison — regularly meets with the chairman and serves as liaison between the chairman and the independent directors; |
| ● | Stockholder communications — makes himself available for direct communication with our stockholders; |
| ● | Board agenda, schedule & information — works with the chairman regarding meeting agendas, meeting schedules and information sent to directors for Board meetings, including the quality, quantity, appropriateness and timeliness of such information; and |
| ● | Advisors and consultants — recommends to the Board the retention of outside advisors and consultants who report directly to the Board on Board-wide issues. |
104
Board Role in Risk Oversight
Risk is an integral part of the Board and Board committee deliberations throughout the year. While the Board has the ultimate oversight responsibility for the risk management process, various committees of the Board also have responsibility for risk management. In particular, the Audit Committee focuses on financial risk, including internal controls, and receives financial risk assessment reports from management. Risks related to the compensation programs are reviewed by the Compensation Committee. The Board is advised by these committees of significant risks and management’s response through periodic updates.
Stockholder Communications with the Board
The Company’s stockholders may communicate with the Board, including non-executive directors or officers, by sending written communications addressed to such person or persons in care of Tonix Pharmaceuticals Holding Corp., Attention: Secretary, 200 Connell Drive, Suite 3100, Berkeley Heights, New Jersey 07922. All communications will be compiled by the Secretary and submitted to the addressee. If the Board modifies this process, the revised process will be posted on the Company’s website.
Meetings and Committees of the Board
During the fiscal year ended December 31, 2025, the Board held 10 meetings, the Audit Committee held eight meetings, the Compensation Committee held eight meetings and the Nominating and Corporate Governance Committee held five meetings. The Board and Board committees also approved certain actions by unanimous written consent.
Each of the directors attended at least 75% of the aggregate of the total number of meetings of our Board (held during the period for which such directors served on the Board). Each of the directors attended at least 75% of the total number of meetings of all committees of our Board on which the director served (during the periods for which the director served on such committee or committees). Dr. Lederman was the only member of the Board who attended last year’s annual meeting of stockholders. The Company does not have a formal policy requiring members of the Board to attend our annual meetings.
Board Committees
The Board has standing Audit, Compensation, and Nominating and Corporate Governance Committees. Information concerning the membership and function of each committee is as follows:
Board Committee Membership
| Name | Audit Committee | Compensation
Committee |
Nominating
and Corporate Governance Committee |
||||
| Richard Bagger | * | ** | |||||
| Margaret Smith Bell | * | ** | |||||
| David Grange | * | * | |||||
| Adeoye Olukotun | * | ||||||
| Newcomb Stillwell | * | * | |||||
| Carolyn Taylor | * | ||||||
| James Treco | ** | * |
| * | Member of Committee |
| ** | Chair of Committee |
Audit Committee
Our Audit Committee consists of James Treco, Chair of the Committee, Richard Bagger, Margaret Smith Bell and Newcomb Stillwell. Our Board has determined each of the members are “independent” as that term is defined under applicable SEC rules and under the current listing standards of the NASDAQ Stock Market. Mr. Treco is our audit committee financial expert.
Our Audit Committee’s responsibilities include: (i) reviewing the independence, qualifications, services, fees, and performance of the independent auditors, (ii) appointing, replacing and discharging the independent auditor, (iii) pre-approving the professional services provided by the independent auditor, (iv) reviewing the scope of the annual audit and reports and recommendations submitted by the independent auditor, and (v) reviewing our financial reporting and accounting policies, including any significant changes, with management and the independent auditor. The Audit Committee reviewed and discussed with management the Company’s audited financial statements for the year ended December 31, 2025. Our Board has adopted a written charter for the Audit Committee, a copy of which is posted under the “Investors” tab under “Governance” on our website, which is located at www.tonixpharma.com.
105
Compensation Committee
Our Compensation Committee consists of Margaret Smith Bell, Chair of the Committee, David Grange, Adeoye Olukotun and Carolyn Taylor. Our Board has determined that all of the members are “independent” under the current listing standards of the NASDAQ Stock Market. Our Board has adopted a written charter setting forth the authority and responsibilities of the Compensation Committee.
Our Compensation Committee has responsibility for, among other things, evaluating and making decisions regarding the compensation of our executive officers, assuring that the executive officers are compensated effectively in a manner consistent with our stated compensation strategy, producing an annual report on executive compensation in accordance with the rules and regulations promulgated by the SEC and periodically evaluating and administering the terms and administration of our incentive plans and benefit programs. In addition, our Compensation Committee reviews and makes recommendations to the Board regarding incentive compensation plans that require shareholder approval, director compensation and the related executive compensation information for inclusion in the Company’s Annual Report on Form 10-K and proxy statement, and employment and severance agreements relating to the chief executive officer. Our Compensation Committee has engaged Aon plc, an independent executive compensation consultant, to provide guidance with respect to the development and implementation of our compensation programs. Our Board has adopted a written charter for the Compensation Committee, a copy of which is posted under the “Investors” tab under “Governance” on our website, which is located at www.tonixpharma.com.
Nominating and Corporate Governance Committee
Our Nominating and Corporate Governance Committee consists of Richard Bagger, Chair of the Committee, David Grange, Newcomb Stillwell and James Treco. The Board has determined that all of the members are “independent” under the current listing standards of the NASDAQ Stock Market.
Our Nominating and Corporate Governance Committee has responsibility for assisting the Board in, among other things, effecting the organization, membership and function of the Board and its committees. The Nominating and Corporate Governance Committee identifies and evaluates the qualifications of all candidates for nomination for election as directors, and seeks director nominees that complement and enhance the effectiveness of the existing Board to ensure that its members have varied and relevant backgrounds, skills, knowledge, perspectives and experiences. In addition, the Nominating and Corporate Governance Committee is responsible for developing, recommending and evaluating corporate governance standards and a code of business conduct and ethics. The Nominating and Corporate Governance Committee is also responsible for developing, recommending and evaluating corporate governance standards and a code of business conduct and ethics. Our Board has adopted a written charter for the Nominating and Corporate Governance Committee, a copy of which is posted under the “Investors” tab under “Governance” on our website, which is located at www.tonixpharma.com.
Nomination of Directors
As provided in its charter and our Company’s corporate governance principles, the Nominating and Corporate Governance Committee is responsible for identifying individuals qualified to become directors. The Nominating and Corporate Governance Committee seeks to identify director candidates based on input provided by a number of sources, including (1) the Nominating and Corporate Governance Committee members, (2) our other directors, (3) our shareholders, (4) our Chief Executive Officer or Chairman, and (5) third parties such as professional search firms. In evaluating potential candidates for director, the Nominating and Corporate Governance Committee considers the entirety of each candidate’s credentials.
Qualifications for consideration as a director nominee may vary according to the particular areas of expertise being sought as a complement to the existing composition of the Board. However, at a minimum, candidates for director must possess:
| ● | high personal and professional ethics and integrity; | |
| ● | the ability to exercise sound judgment; |
106
| ● | the ability to make independent analytical inquiries; | |
| ● | a willingness and ability to devote adequate time and resources to diligently perform Board and committee duties; and | |
| ● | the appropriate and relevant business experience and acumen. |
In addition to these minimum qualifications, the Nominating and Corporate Governance Committee also takes into account when considering whether to nominate a potential director candidate the following factors:
| ● | whether the person possesses specific industry expertise and familiarity with general issues affecting our business; | |
| ● | whether the person’s nomination and election would enable the Board to have a member that qualifies as an “audit committee financial expert” as such term is defined by the SEC in Item 401 of Regulation S-K; | |
| ● | whether the person would qualify as an “independent” director under the listing standards of the Nasdaq Stock Market; | |
| ● | the importance of continuity of the existing composition of the Board to provide long term stability and experienced oversight; and | |
| ● | the importance of diversified Board membership, in terms of both the individuals involved and their various experiences and areas of expertise. |
The Nominating and Corporate Governance Committee will consider director candidates recommended by shareholders provided such recommendations are submitted in accordance with the procedures set forth below. In order to provide for an orderly and informed review and selection process for director candidates, the Board has determined that shareholders who wish to recommend director candidates for consideration by the Nominating and Corporate Governance Committee must comply with the following:
| ● | The recommendation must be made in writing to the Corporate Secretary at Tonix Pharmaceuticals Holding Corp.; | |
| ● | The recommendation must include the candidate’s name, home and business contact information, detailed biographical data and qualifications, information regarding any relationships between the candidate and the Company within the last three years and evidence of the recommending person’s ownership of the Company’s common stock; | |
| ● | The recommendation shall also contain a statement from the recommending shareholder in support of the candidate; professional references, particularly within the context of those relevant to board membership, including issues of character, judgment, diversity, age, independence, expertise, corporate experience, length of service, other commitments and the like; and personal references; and | |
| ● | A statement from the shareholder nominee indicating that such nominee wants to serve on the Board and could be considered “independent” under the Rules and Regulations of the Nasdaq Stock Market and the SEC, as in effect at that time. |
All candidates submitted by shareholders will be evaluated by the Nominating and Corporate Governance Committee according to the criteria discussed above and in the same manner as all other director candidates.
Prohibition Against Certain Transactions
All of our employees and directors are prohibited from hedging or pledging Tonix stock, or engaging in short sales or trading in standardized options under our Statement of Company Policy on Insider Trading and Policy Regarding Special Trading Procedures (the “Insider Trading Policy”).
107
Insider Trading Policies and Procedures
Code of Ethics
We have adopted a Code of Business Conduct and Ethics that applies to all of our directors, officers and employees which can be found on our website at https://ir.tonixpharma.com/corporate-governance/governance-documents. We intend to disclose future amendments to certain provisions of our Code of Business Conduct and Ethics, or waivers of such provisions applicable to any principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions, and our directors, on our website identified above or in filings with the SEC.
Delinquent Section 16(a) Reports
Section 16(a) of the Exchange Act requires our directors, executive officers and persons who beneficially own more than 10% of our outstanding common shares to file reports with the SEC regarding their share ownership and changes in their ownership of our common shares. Based on our records and representations from our directors and executive officers, we believe that all Section 16(a) filing requirements applicable to our directors and executive officers were complied with during the fiscal year ended December 31, 2025.
ITEM 11 – EXECUTIVE COMPENSATION
Compensation Philosophy and Practices
We believe that the performance of our executive officers significantly impacts our ability to achieve our corporate goals. We, therefore, place considerable importance on the design and administration of our executive officer compensation program. This program is intended to enhance stockholder value by attracting, motivating and retaining qualified individuals to perform at the highest levels and to contribute to our growth and success. Our executive officer compensation program is designed to provide compensation opportunities that are tied to individual and corporate performance.
Our compensation packages are also designed to be competitive in our industry. The Compensation Committee from time-to-time consults with compensation consultants, legal counsel and other advisors in designing our compensation program, including in evaluating the competitiveness of individual compensation packages and in relation to our corporate goals.
108
Our overall compensation philosophy has been to pay our executive officers an annual base salary and to provide opportunities, through cash and equity incentives, to provide higher compensation if certain key performance goals are satisfied. We believe that many of our key practices and programs demonstrate good governance. The main principles of our fiscal year 2024 compensation strategy included the following:
| ● | An emphasis on pay for performance. A significant portion of our executive officers’ total compensation is variable and at risk and tied directly to measurable performance, including pre-specified corporate, strategic or developmental goals, which aligns the interests of our executives with those of our stockholders; |
| ● | Performance results are linked to Company and individual performance. When looking at performance over the year, we equally weigh individual performance as well as that of the Company as a whole. Target annual compensation is positioned to allow for above-median compensation to be earned through an executive officer’s and the Company’s extraordinary performance; |
| ● | Equity as a key component to align the interests of our executives with those of our stockholders. Our Compensation Committee believes that keeping executives interests aligned with those of our stockholders is critical to driving toward achievement of long-term goals of both our stockholders and the Company. Accordingly, a significant portion of our executives’ compensation are stock based, including stock options that are exercisable at a percentage above market value at the time of grant; and |
| ● | Peer group positioning. While the Compensation Committee considers the level of compensation paid by the companies in our peer group as a reference point that provides a framework for its compensation decisions, in order to maintain competitiveness and flexibility, the Compensation Committee does not employ a formal benchmarking strategy or rely upon specific peer–derived targets. |
In 2025, we also continued practices that demonstrate good governance and careful stewardship of corporate assets, including:
| ● | Limited personal benefits. Our executive officers are eligible for the same benefits as our non-executive salaried employees, and they do not receive any additional perquisites. |
| ● | No retirement benefits. We do not provide our executive officers with a traditional retirement plan, or with any supplemental deferred compensation or retirement benefits. | |
| ● | No tax gross-ups. We do not provide our executive officers with any tax gross-ups. | |
| ● | No single-trigger cash change in control benefits. We do not provide cash benefits to, or accelerate the vesting of unvested equity grants issued to, our executives upon a change in control, absent an actual termination of employment. |
At our annual meeting in May 2025, we conducted our tri-annual advisory vote on executive compensation, commonly referred to as a “say-on-pay” vote. At that time, a majority of the votes affirmatively cast on the advisory say-on-pay proposal were voted in favor of the compensation of our named executive officers. The Compensation Committee understood this level of approval to indicate strong stockholder support for our executive compensation policies and programs generally, and as a result, our Compensation Committee made no fundamental changes to our executive compensation programs. We will hold our next say-on-pay vote at the 2028 annual meeting. Our Compensation Committee and our Board will consider shareholder feedback through the say-on-pay vote and remains committed to engaging with shareholders and are open to feedback from shareholders.
Summary Compensation Table
The following table provides certain summary information concerning compensation awarded to, earned by or paid to our Chief Executive Officer, and the two next most highly paid executive officers for fiscal years 2025 and 2024.
| Name
& Principal Position |
Year | Salary
($) |
Bonus
($) |
Stock
Awards ($) |
Option
Awards ($)(1) |
Non-Equity
Incentive Plan Compensation ($) |
All
Other Compensation ($) |
Total ($) |
||||||||||||||||||||||||||||||||||
| Seth Lederman | 2025 | 702,000 | 596,700 | — | 1,870,130 | — | — | 3,168,830 | ||||||||||||||||||||||||||||||||||
| Chief Executive Officer | 2024 | 675,000 | 417,656 | — | 717,111 | — | — | 1,809,767 | ||||||||||||||||||||||||||||||||||
| Jessica Morris | 2025 | 522,912 | 261,450 | — | 441,907 | — | — | 1,226,269 | ||||||||||||||||||||||||||||||||||
| Chief Operations Officer | 2024 | 494,000 | 180,310 | — | 213,431 | — | — | 887,741 | ||||||||||||||||||||||||||||||||||
| Bradley Saenger | 2025 | 502,944 | 251,450 | — | 389,119 | — | — | 1,143,513 | ||||||||||||||||||||||||||||||||||
| Chief Financial Officer | 2024 | 483,600 | 176,514 | — | 189,867 | — | — | 849,981 | ||||||||||||||||||||||||||||||||||
| Gregory Sullivan | 2025 | 519,168 | 259,600 | — | 437,774 | — | — | 1,216,542 | ||||||||||||||||||||||||||||||||||
| Chief Medical Officer | 2024 | 499,200 | 182,208 | — | 241,036 | — | — | 922,444 | ||||||||||||||||||||||||||||||||||
| (1) | Represents the aggregate grant date fair value of options granted in accordance with Financial Accounting Standards Board, or FASB, Accounting Standards Codification, or ASC, Topic 718, “Stock Compensation.” For the relevant assumptions used in determining these amounts, refer to Note 18 to our audited financial statements. |
109
Grants of Plan-Based Awards in Fiscal 2025
The following table provides information with regard to each grant of plan-based award made to a named executive officer under any plan during the fiscal year ended December 31, 2025.
| Name | Grant Date | All
Other Option Awards: Number of Securities Underlying Options(#) |
Exercise
or Base Price of Option Awards ($/Share) |
Grant
Date Fair Value of Stock and Option Awards ($)(1) |
|||||||||||||
| Seth Lederman | 2/25/2025 | 123,958 | 8.05 | 938,610 | |||||||||||||
| 2/25/2025 | 123,958 | 10.06 | (2) | 931,520 | |||||||||||||
| Bradley Saenger | 2/25/2025 | 25,792 | 8.05 | 195,297 | |||||||||||||
| 2/25/2025 | 25,792 | 10.06 | (2) | 193,822 | |||||||||||||
| Jessica Morris | 2/25/2025 | 29,291 | 8.05 | 221,791 | |||||||||||||
| 2/25/2025 | 29,291 | 10.06 | (2) | 220,116 | |||||||||||||
| Gregory Sullivan | 2/25/2025 | 29,017 | 8.05 | 219,717 | |||||||||||||
| 2/25/2025 | 29,017 | 10.06 | (2) | 218,057 | |||||||||||||
| (1) | Represents the aggregate grant date fair value of options granted in accordance with FASB ASC Topic 718. |
| (2) | Represents an exercise price at a 125% premium of the closing price of the Company’s common stock on the grant date. |
Policies and Practices Related to the Grant of Certain Equity Awards Close in Time to the Release of Material Nonpublic Information
We do not have any formal policy that requires us to grant, or avoid granting, stock options at particular times. Consistent with our annual compensation cycle, if options are to be granted, the Compensation Committee generally seeks to grant annual stock option awards in connection with their conducting and completing such annual review, which typically occurs in approximately February of each year. Options are awarded to our non-employee directors pursuant to our Amended and Restated 2020 Plan, which is awarded on the date of our annual meeting of stockholders. The timing of any stock option grants in connection with new hires, promotions, or other non-routine grants may be tied to the event giving rise to the award (such as an employee’s commencement of employment or promotion effective date), and in other cases such grants may be awarded at the same time with other annual grants. As a result, in all cases, the timing of grants of stock options occurs independent of the release of any material nonpublic information, and we do not time the disclosure of material nonpublic information for the purpose of affecting the value of executive compensation.
110
No stock options were issued to executive officers in 2025 during any period beginning four business days before the filing of a periodic report or current report disclosing material non-public information (other than a current report on Form 8-K disclosing a material new option award grant under Item 5.02(e) of that form) and ending one business day after the filing or furnishing of such report with the SEC.
Outstanding Equity Awards at December 31, 2025
The following table presents information regarding outstanding equity awards held by our named executive officers as of December 31, 2025.
| Name | Number
of Securities underlying Unexercised Options (#) Exercisable |
Number
of Securities underlying Unexercised Options (#) Unexercisable |
Option
Exercise Price ($/Sh) |
Option Expiration Date | ||||||||||
| Seth Lederman | 1 | — | $ | 3,219,200,000.00 | 2/9/2026 | |||||||||
| — | 1 | (1) | $ | 3,219,200,000.00 | 2/9/2026 | |||||||||
| 1 | — | $ | 352,000,000.00 | 3/1/2027 | ||||||||||
| 1 | — | $ | 217,600,000.00 | 2/13/2028 | ||||||||||
| 1 | — | $ | 272,000,000.00 | 2/13/2028 | ||||||||||
| 1 | — | $ | 12,096,000.00 | 2/26/2029 | ||||||||||
| 1 | — | $ | 15,104,000.00 | 2/26/2029 | ||||||||||
| 1 | — | $ | 13,120,000.00 | 5/6/2029 | ||||||||||
| 1 | — | $ | 16,384,000.00 | 5/6/2029 | ||||||||||
| 1 | — | $ | 256,000.00 | 2/25/2030 | ||||||||||
| 1 | — | $ | 320,000.00 | 2/25/2030 | ||||||||||
| 4 | — | $ | 492,800.00 | 5/4/2030 | ||||||||||
| 4 | — | $ | 614,400.00 | 5/4/2030 | ||||||||||
| 5 | — | $ | 780,800.00 | 2/23/2031 | ||||||||||
| 5 | — | $ | 979,200.00 | 2/23/2031 | ||||||||||
| 8 | — | (2) | $ | 134,400.00 | 2/15/2032 | |||||||||
| 5 | 3 | (2) | $ | 262,400.00 | 2/15/2032 | |||||||||
| 5 | 3 | (2) | $ | 396,800.00 | 2/15/2032 | |||||||||
| 5 | 3 | (2) | $ | 531,200.00 | 2/15/2032 | |||||||||
| 34 | 1 | (3) | $ | 14,624.00 | 2/23/2033 | |||||||||
| 34 | 1 | (3) | $ | 18,208.00 | 2/23/2033 | |||||||||
| 410 | — | (4) | $ | 1,177.60 | 2/27/2034 | |||||||||
| 46 | 28 | (5) | $ | 1,177.60 | 2/27/2034 | |||||||||
| 369 | 226 | (5) | $ | 1,472.00 | 2/27/2034 | |||||||||
| — | 123,958 | (6) | $ | 8.05 | 2/25/2035 | |||||||||
| — | 123,958 | (6) | $ | 10.06 | 2/25/2035 | |||||||||
| Jessica Morris | 1 | — | $ | 3,219,200,000.00 | 2/9/2026 | |||||||||
| — | 1 | (1) | $ | 3,219,200,000.00 | 2/9/2026 | |||||||||
| 1 | — | $ | 352,000,000.00 | 3/1/2027 | ||||||||||
| 1 | — | $ | 217,600,000.00 | 2/13/2028 | ||||||||||
| 1 | — | $ | 272,000,000.00 | 2/13/2028 | ||||||||||
| 1 | — | $ | 12,096,000.00 | 2/26/2029 | ||||||||||
| 1 | — | $ | 15,104,000.00 | 2/26/2029 | ||||||||||
| 1 | — | $ | 13,120,000.00 | 5/6/2029 | ||||||||||
| 1 | — | $ | 16,384,000.00 | 5/6/2029 | ||||||||||
| 1 | — | $ | 256,000.00 | 2/25/2030 | ||||||||||
| 1 | — | $ | 320,000.00 | 2/25/2030 | ||||||||||
| 1 | — | $ | 492,800.00 | 5/4/2030 | ||||||||||
| 1 | — | $ | 614,400.00 | 5/4/2030 | ||||||||||
| 1 | — | $ | 780,800.00 | 2/23/2031 | ||||||||||
| 1 | — | $ | 979,200.00 | 2/23/2031 | ||||||||||
| 2 | — | (2) | $ | 134,400.00 | 2/15/2032 | |||||||||
| 1 | 1 | (2) | $ | 262,400.00 | 2/15/2032 | |||||||||
| 1 | 1 | (2) | $ | 396,800.00 | 2/15/2032 | |||||||||
| 1 | 1 | (2) | $ | 531,200.00 | 2/15/2032 | |||||||||
| 7 | 4 | (3) | $ | 14,624.00 | 2/23/2033 | |||||||||
| 7 | 4 | (3) | $ | 18,272.00 | 2/23/2033 | |||||||||
| 63 | 37 | (5) | $ | 1,177.60 | 2/27/2034 | |||||||||
| 63 | 37 | (5) | $ | 1,472.00 | 2/27/2034 | |||||||||
| — | 29,291 | (6) | $ | 8.05 | 2/25/2035 | |||||||||
| — | 29,291 | (6) | $ | 10.06 | 2/25/2035 | |||||||||
| Bradley Saenger | 1 | — | $ | 3,219,200,000.00 | 2/9/2026 | |||||||||
| — | 1 | (1) | $ | 1,548,800,000.00 | 5/27/2026 | |||||||||
| 1 | — | $ | 1,548,800,000.00 | 5/27/2026 | ||||||||||
| 1 | — | $ | 352,000,000.00 | 3/1/2027 | ||||||||||
| 1 | — | $ | 217,600,000.00 | 2/13/2028 | ||||||||||
| 1 | — | $ | 272,000,000.00 | 2/13/2028 | ||||||||||
| 1 | — | $ | 12,096,000.00 | 2/26/2029 | ||||||||||
| 1 | — | $ | 15,104,000.00 | 2/26/2029 | ||||||||||
| 1 | — | $ | 13,120,000.00 | 5/6/2029 | ||||||||||
| 1 | — | $ | 16,384,000.00 | 5/6/2029 | ||||||||||
| 1 | — | $ | 256,000.00 | 2/25/2030 | ||||||||||
| 1 | — | $ | 320,000.00 | 2/25/2030 | ||||||||||
| 1 | — | $ | 492,800.00 | 5/4/2030 | ||||||||||
| 1 | — | $ | 614,400.00 | 5/4/2030 | ||||||||||
| 1 | — | $ | 780,800.00 | 2/23/2031 | ||||||||||
| 1 | — | $ | 979,200.00 | 2/23/2031 | ||||||||||
| 1 | 1 | (2) | $ | 134,400.00 | 2/15/2032 | |||||||||
| 1 | 1 | (2) | $ | 262,400.00 | 2/15/2032 | |||||||||
| 1 | 1 | (2) | $ | 396,800.00 | 2/15/2032 | |||||||||
| 1 | 1 | (2) | $ | 531,200.00 | 2/15/2032 | |||||||||
| 7 | 3 | (3) | $ | 14,624.00 | 2/23/2033 | |||||||||
| 7 | 3 | (3) | $ | 18,272.00 | 2/23/2033 | |||||||||
| 60 | 29 | (5) | $ | 1,177.60 | 2/27/2034 | |||||||||
| 60 | 29 | (5) | $ | 1,472.00 | 2/27/2034 | |||||||||
| — | 25,792 | (6) | $ | 8.05 | 2/25/2035 | |||||||||
| — | 25,792 | (6) | $ | 10.06 | 2/25/2035 | |||||||||
| Gregory Sullivan | 1 | — | $ | 3,219,200,000.00 | 2/9/2026 | |||||||||
| — | 1 | (1) | $ | 3,219,200,000.00 | 2/9/2026 | |||||||||
| 1 | — | $ | 352,000,000.00 | 3/1/2027 | ||||||||||
| 1 | — | $ | 217,600,000.00 | 2/13/2028 | ||||||||||
| 1 | — | $ | 272,000,000.00 | 2/13/2028 | ||||||||||
| 1 | — | $ | 12,096,000.00 | 2/26/2029 | ||||||||||
| 1 | — | $ | 15,104,000.00 | 2/26/2029 | ||||||||||
| 1 | — | $ | 13,120,000.00 | 5/6/2029 | ||||||||||
| 1 | — | $ | 16,384,000.00 | 5/6/2029 | ||||||||||
| 1 | — | $ | 256,000.00 | 2/25/2030 | ||||||||||
| 1 | — | $ | 320,000.00 | 2/25/2030 | ||||||||||
| 1 | — | $ | 492,800.00 | 5/4/2030 | ||||||||||
| 1 | — | $ | 614,400.00 | 5/4/2030 | ||||||||||
| 1 | — | $ | 780,800.00 | 2/23/2031 | ||||||||||
| 1 | — | $ | 979,200.00 | 2/23/2031 | ||||||||||
| 2 | — | (2) | $ | 134,400.00 | 2/15/2032 | |||||||||
| 1 | 1 | (2) | $ | 262,400.00 | 2/15/2032 | |||||||||
| 1 | 1 | (2) | $ | 396,800.00 | 2/15/2032 | |||||||||
| 1 | 1 | (2) | $ | 531,200.00 | 2/15/2032 | |||||||||
| 8 | 3 | (3) | $ | 14,624.00 | 2/23/2033 | |||||||||
| 8 | 3 | (3) | $ | 18,272.00 | 2/23/2033 | |||||||||
| 70 | 42 | (5) | $ | 1,177.60 | 2/27/2034 | |||||||||
| 70 | 42 | (5) | $ | 1,472.00 | 2/27/2034 | |||||||||
| — | 29,017 | (6) | $ | 8.05 | 2/25/2035 | |||||||||
| — | 29,017 | (6) | $ | 10.06 | 2/25/2035 |
| (1) | The shares subject to this stock option vest 1/3rd upon the date(s) that certain stock price goals are achieved. The stock price goals are such date(s) when the Company’s common stock has an average closing sales price equal to or exceeding each of $3,840,000,000.00, $4,480,000,000.00 and $5,120,000,000.00 per share for 20 consecutive trading days, subject to a one year minimum service period prior to vesting. |
| (2) | The shares subject to this stock option vested as to 10% of the shares on February 15, 2023, 10% of the shares on February 15, 2024, 40% of the shares on February 15, 2025 and 40% of the shares on February 15, 2026. |
| (3) | The shares subject to this stock option vested as to 1/3rd of the shares on February 23, 2024, with the remaining shares vesting on an equal monthly basis over the following 24 months. |
| (4) | The shares subject to this stock option were in lieu of a cash award, Dr. Lederman’s 2024 cash bonus was paid in the form of a stock option award granted pursuant to the 2020 Plan, with 100% of such options vesting on the six-month anniversary of issuance, expiring 10 years from date of issuance. |
| (5) | The shares subject to this stock option vested as to 1/3 of the shares on February 23, 2025, with the remaining shares vesting on an equal monthly basis over the following 24 months. |
| (6) | The shares subject to this stock option vested as to 1/3 of the shares on February 25, 2026, with the remaining shares vesting on an equal monthly basis over the following 24 months.
|
111
Option Exercises and Stock Vested
No options were exercised by any of the named executive officers and no named executive officers held restricted stock units during the fiscal year ended December 31, 2025.
Equity Compensation Plan Information
The following table provides certain information with respect to our equity compensation plans in effect as of December 31, 2025.
| Plan Category | Number
of securities to be issued upon exercise of outstanding options, warrants and rights (A) |
Weighted- average exercise price of outstanding options, warrants and rights (B) |
Number
of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column A)(2) (C) | |||||||||
| Equity compensation plans approved by security holders(1) | 1,150,551 | $ | 43,825.73 | 2,000,159 | ||||||||
| Equity compensation plans not approved by security holders | — | — | — | |||||||||
| Total | 1,150,551 | $ | 43,825.73 | 2,000,159 | ||||||||
| (1) | Consists of the Company’s 2014 Stock Incentive Plan, the 2016 Stock Incentive Plan, the 2017 Stock Incentive Plan, the 2018 Equity Incentive Plan, the 2019 Stock Incentive Plan, the 2020 Stock Incentive Plan, the Amended and Restated 2020 Stock Incentive Plan and the 2019 Employee Stock Purchase Plan, the 2020 Employee Stock Purchase Plan, 2022 Employee Stock Purchase Plan, and the 2025 Employee Stock Purchase Plan (the “ESPP”). |
| (2) | Consists of shares available for future issuance under the Amended and Restated 2020 Plan and our ESPP. As of December 31, 2025, 159 shares of common stock were available for issuance under the Amended and Restated 2023 Plan and 2,000,000 shares of common stock were available for issuance under the 2025 ESPP. |
112
Employment Contracts and Termination of Employment and Change-In-Control Arrangements
Employment Agreement with Seth Lederman
On February 11, 2014, the Company entered into an employment agreement (the “Lederman Agreement”) with Dr. Seth Lederman to continue to serve as our President, Chief Executive Officer and Chairman of the Board.
The base salary for Dr. Lederman under the Lederman Agreement was $425,000 per annum and as of March 1, 2026, the base salary is $743,000. The Lederman Agreement has an initial term of one year and automatically renews for successive one year terms unless either party delivers written notice not to renew at least 60 days prior to the end of the current term.
Pursuant to the Lederman Agreement, if the Company terminates Dr. Lederman’s employment without Cause (as defined in the Lederman Agreement) or Dr. Lederman resigns for Good Reason (as defined in the Lederman Agreement), Dr. Lederman is entitled to the following payments and benefits: (1) his fully earned but unpaid base salary through the date of termination at the rate then in effect, plus all other benefits, if any, under any group retirement plan, nonqualified deferred compensation plan, equity award plan or agreement, health benefits plan or other group benefit plan to which Dr. Lederman may be entitled to under the terms of such plans or agreements; (2) a lump sum cash payment in an amount equal to 12 months of his base salary as in effect immediately prior to the date of termination; (3) continuation of health benefits for Dr. Lederman and his eligible dependents for a period of 12 months following the date of termination; and (4) the automatic acceleration of the vesting and exercisability of outstanding unvested stock awards as to the number of stock awards that would have vested over the 12-month period following termination had Dr. Lederman remained continuously employed by the Company during such period.
Pursuant to the Lederman Agreement, if Dr. Lederman’s employment is terminated as a result of death or permanent disability, Dr. Lederman or his estate, as applicable, is entitled to the following payments and benefits: (1) his fully earned but unpaid base salary through the date of termination at the rate then in effect; (2) a lump sum cash payment in an amount equal to six months of his base salary as in effect immediately prior to the date of termination; and (3) the automatic acceleration of the vesting and exercisability of outstanding unvested stock awards.
If Dr. Lederman is terminated without Cause or resigns for Good Reason during the period commencing 90 days prior to a Change in Control (as defined below) or 12 months following a Change in Control, Dr. Lederman shall be entitled to receive, in lieu of the severance benefits described above, the following payments and benefits: (1) a lump sum cash payment in an amount equal to 36 months of his base salary as in effect immediately prior to the date of termination, except that, if and while Dr. Lederman is still entitled to the Sale Bonus (as defined below), it will only be 18 months; (2) continuation of health benefits for Dr. Lederman and his eligible dependents for a period of 24 months following the date of termination, except that, if and while Dr. Lederman is still entitled to the Sale Bonus it will only be 12 months; and (3) the automatic acceleration of the vesting and exercisability of outstanding unvested stock awards.
If during the term of the Lederman Agreement or within 120 days after Dr. Lederman is terminated without Cause or resigns for Good Reason, following a Change in Control, the Company consummates a Change in Control transaction in which the Enterprise Value (as defined below) equals or exceeds $50 million, Dr. Lederman shall be entitled to receive a lump sum payment equal to 4.4% of the Enterprise Value (the “Sale Bonus”). The Sale Bonus provision of the Lederman Agreement will terminate upon the Company granting Dr. Lederman long-term incentive compensation mutually agreed to by the Board and Dr. Lederman.
For purposes of the Lederman Agreement, “Cause” generally means (1) commission of an act of fraud, embezzlement or dishonesty or some other illegal act that has a demonstrable material adverse impact on the Company or any successor or affiliate of the Company, (2) conviction of, or entry into a plea of “guilty” or “no contest” to, a felony, (3) unauthorized use or disclosure of the Company’s confidential information or trade secrets or any successor or affiliate of the Company that has, or may reasonably be expected to have, a material adverse impact on any such entity; (4) gross negligence, failure to follow a material, lawful and reasonable request of the Board or material violation of any duty of loyalty to the Company or any successor or affiliate of the Company, or any other demonstrable material willful misconduct by Dr. Lederman, (5) ongoing and repeated failure or refusal to perform or neglect of his duties as required by his employment agreement, which failure, refusal or neglect continues for 30 days following Dr. Lederman’s receipt of written notice from the Board stating with specificity the nature of such failure, refusal or neglect, provided that such failure to perform is not as a result of illness, injury or medical incapacity, or (6) material breach of any Company policy or any material provision of the Lederman Agreement.
113
For purposes of the Lederman Agreement, “Good Reason” generally means (1) a material diminution in Dr. Lederman’s title, authority, duties or responsibilities, (2) a material diminution in Dr. Lederman’s base compensation, unless such a reduction is imposed across-the-board to the Company’s senior management, and such reduction is not greater than 15%, (3) a material change in the geographic location at which Dr. Lederman must perform his duties, (4) any other action or inaction that constitutes a material breach by the Company or any successor or affiliate of the Company’s obligations to Dr. Lederman under the Lederman Agreement, or (5) the Company elects not to renew the Lederman Agreement for another term.
For purposes of the Lederman Agreement, “Change in Control” generally means:
| ● | A transaction or series of transactions (other than public offerings) that results in any person or entity or related group of persons or entities (other than the Company, its subsidiaries, an employee benefit plan maintained by the Company or any of its subsidiaries or a person or entity that, prior to such transaction, directly or indirectly controls, is controlled by, or is under common control with, the Company) of beneficial ownership (within the meaning of Rule 13d-3 under the Exchange Act) of more than 40% of the total combined voting power of the Company’s securities outstanding immediately after such acquisition; |
| ● | (1) a merger, consolidation, reorganization, or business combination or (2) the sale, exchange or transfer of all or substantially all of the Company’s assets in any single transaction or series of transactions or (3) the acquisition of assets or stock of another entity, in each case other than a transaction: |
| ○ | which results in the Company’s voting securities outstanding immediately before the transaction continuing to represent, directly or indirectly, at least 60% of the combined voting power of the successor entity’s outstanding voting securities immediately after the transaction, and | |
| ○ | after which no person or group beneficially owns voting securities representing 40% or more of the combined voting power of the Company or its successor; provided, however, that no person or group is treated as beneficially owning 40% or more of combined voting power of the Company or its successor solely as a result of the voting power held in the Company prior to the consummation of the transaction. |
For purposes of the Lederman Agreement, “Enterprise Value” generally means (1) in a Change in Control in which consideration is received by the Company, the total cash and non-cash consideration, including debt assumed, received by the Company, net of any fees and expenses in connection with the transaction and (2) in a Change in Control in which consideration is payable to the stockholders of the Company, the total cash and non-cash consideration, including debt assumed, payable to the Company’s stockholders net of any fees and expenses in connection with the transaction. Enterprise Value also includes any cash or non-cash consideration payable to the Company or to the Company’s stockholders on a contingent, earnout or deferred basis.
Employment Agreement with Gregory Sullivan
On June 3, 2014, the Company entered into an employment agreement (the “Sullivan Agreement”) with Dr. Gregory Sullivan to serve as our Chief Medical Officer. The base salary for Dr. Sullivan under the Sullivan Agreement was $225,000 per annum and as of March 1, 2026, the base salary is $550,000. The Sullivan Agreement had an initial term of one year and automatically renews for successive one year terms unless either party delivers written notice not to renew at least 60 days prior to the end of the current term.
Pursuant to the Sullivan Agreement, if the Company terminates Dr. Sullivan’s employment without Cause (as defined below) or Executive resigns for Good Reason (as defined below), Dr. Sullivan is entitled to the following payments and benefits: (1) his fully earned but unpaid base salary through the date of termination at the rate then in effect, plus all other benefits, if any, under any group retirement plan, nonqualified deferred compensation plan, equity award plan or agreement, health benefits plan or other group benefit plan to which Dr. Sullivan may be entitled to under the terms of such plans or agreements; (2) a lump sum cash payment in an amount equal to 12 months of his base salary as in effect immediately prior to the date of termination; (3) continuation of health benefits for Dr. Sullivan and his eligible dependents for a period of 12 months following the date of termination; and (4) the automatic acceleration of the vesting and exercisability of outstanding unvested stock awards as to the number of stock awards that would have vested over the 12-month period following termination had Dr. Sullivan remained continuously employed by the Company during such period.
Pursuant to the Sullivan Agreement, if Dr. Sullivan’s employment is terminated as a result of death or permanent disability, Dr. Sullivan or his estate, as applicable, is entitled to his fully earned but unpaid base salary through the end of the month in which termination occurs at the rate then in effect.
114
For purposes of the Sullivan Agreement, “Cause” generally means (1) commission of an act of fraud, embezzlement or dishonesty or some other illegal act that has a demonstrable material adverse impact on the Company or any successor or affiliate of the Company, (2) conviction of, or entry into a plea of “guilty” or “no contest” to, a felony, (3) unauthorized use or disclosure of the Company’s confidential information or trade secrets or any successor or affiliate of the Company that has, or may reasonably be expected to have, a material adverse impact on any such entity, (4) gross negligence, failure to follow a material, lawful and reasonable request of the Company or material violation of any duty of loyalty to the Company or any successor or affiliate of the Company, or any other demonstrable material misconduct by Dr. Sullivan, (5) ongoing and repeated failure or refusal to perform or neglect of his duties as required by his employment agreement, which failure, refusal or neglect continues for 30 days following Dr. Sullivan’s receipt of written notice from the Company stating with specificity the nature of such failure, refusal or neglect, or (6) material breach of any Company policy or any material provision of the Sullivan Agreement.
For purposes of the Sullivan Agreement, “Good Reason” generally means (1) a material diminution in Dr. Sullivan’s title, authority, duties or responsibilities, (2) a material diminution in the executive officer’s base compensation, unless such a reduction is imposed across-the-board to the Company’s senior management and such reduction is not greater than 15%, (3) a material change in the geographic location at which the executive officer must perform his duties, (4) any other action or inaction that constitutes a material breach by the Company or any successor or affiliate of the Company’s obligations to Dr. Sullivan under the Agreement, or (5) the Company elects not to renew the Agreement for another term.
Employment Agreement with Bradley Saenger
On February 23, 2021, the Company entered into an employment agreement (the “Saenger Agreement”) with Mr. Bradley Saenger to serve as our Chief Financial Officer. The base salary for Mr. Saenger under the Saenger Agreement was $545,000 per annum as of March 1, 2026. The Saenger Agreement has an initial term of one year and automatically renews for successive one year terms unless either party delivers written notice not to renew at least 60 days prior to the end of the current term.
Pursuant to the Saenger Agreement, if the Company terminates Mr. Saenger’s employment without Cause (as defined below) or Executive resigns for Good Reason (as defined below), Mr. Saenger is entitled to the following payments and benefits: (1) his fully earned but unpaid base salary through the date of termination at the rate then in effect, plus all other benefits, if any, under any group retirement plan, nonqualified deferred compensation plan, equity award plan or agreement, health benefits plan or other group benefit plan to which Mr. Saenger may be entitled to under the terms of such plans or agreements; (2) a lump sum cash payment in an amount equal to 12 months of his base salary as in effect immediately prior to the date of termination; (3) continuation of health benefits for Mr. Saenger and his eligible dependents for a period of 12 months following the date of termination; and (4) the automatic acceleration of the vesting and exercisability of outstanding unvested stock awards as to the number of stock awards that would have vested over the 12-month period following termination had Mr. Saenger remained continuously employed by the Company during such period.
Pursuant to the Saenger Agreement, if Mr. Saenger’s employment is terminated as a result of death or permanent disability, Mr. Saenger or his estate, as applicable, is entitled to his fully earned but unpaid base salary through the end of the month in which termination occurs at the rate then in effect.
For purposes of the Saenger Agreement, “Cause” generally means (1) commission of an act of fraud, embezzlement or dishonesty or some other illegal act that has a demonstrable material adverse impact on the Company or any successor or affiliate of the Company, (2) conviction of, or entry into a plea of “guilty” or “no contest” to, a felony, (3) unauthorized use or disclosure of the Company’s confidential information or trade secrets or any successor or affiliate of the Company that has, or may reasonably be expected to have, a material adverse impact on any such entity, (4) gross negligence, failure to follow a material, lawful and reasonable request of the Company or material violation of any duty of loyalty to the Company or any successor or affiliate of the Company, or any other demonstrable material misconduct by Mr. Saenger, (5) ongoing and repeated failure or refusal to perform or neglect of his duties as required by his employment agreement, which failure, refusal or neglect continues for 30 days following Mr. Saenger’s receipt of written notice from the Company stating with specificity the nature of such failure, refusal or neglect, or (6) material breach of any Company policy or any material provision of the Saenger Agreement.
For purposes of the Saenger Agreement, “Good Reason” generally means (1) a material diminution in Mr. Saenger’s title, authority, duties or responsibilities, (2) a material diminution in the executive officer’s base compensation, unless such a reduction is imposed across-the-board to the Company’s senior management and such reduction is not greater than 15%, (3) a material change in the geographic location at which the executive officer must perform his duties, (4) any other action or inaction that constitutes a material breach by the Company or any successor or affiliate of the Company’s obligations to Mr. Saenger under the Agreement, or (5) the Company elects not to renew the Agreement for another term.
Employment Agreement with Jessica Morris
On February 23, 2021, the Company entered into an employment agreement (the “Morris Agreement”) with Ms. Jessica Morris to serve as our Chief Operating Officer. The base salary for Ms. Morris under the Morris Agreement was $580,000 per annum as of March 1, 2026. The Morris Agreement has an initial term of one year and automatically renews for successive one year terms unless either party delivers written notice not to renew at least 60 days prior to the end of the current term.
115
Pursuant to the Morris Agreement, if the Company terminates Ms. Morris’s employment without Cause (as defined below) or Executive resigns for Good Reason (as defined below), Ms. Morris is entitled to the following payments and benefits: (1) her fully earned but unpaid base salary through the date of termination at the rate then in effect, plus all other benefits, if any, under any group retirement plan, nonqualified deferred compensation plan, equity award plan or agreement, health benefits plan or other group benefit plan to which Ms. Morris may be entitled to under the terms of such plans or agreements; (2) a lump sum cash payment in an amount equal to 12 months of her base salary as in effect immediately prior to the date of termination; (3) continuation of health benefits for Ms. Morris and her eligible dependents for a period of 12 months following the date of termination; and (4) the automatic acceleration of the vesting and exercisability of outstanding unvested stock awards as to the number of stock awards that would have vested over the 12-month period following termination had Ms. Morris remained continuously employed by the Company during such period.
Pursuant to the Morris Agreement, if Ms. Morris’s employment is terminated as a result of death or permanent disability, Ms. Morris or her estate, as applicable, is entitled to her fully earned but unpaid base salary through the end of the month in which termination occurs at the rate then in effect.
For purposes of the Morris Agreement, “Cause” generally means (1) commission of an act of fraud, embezzlement or dishonesty or some other illegal act that has a demonstrable material adverse impact on the Company or any successor or affiliate of the Company, (2) conviction of, or entry into a plea of “guilty” or “no contest” to, a felony, (3) unauthorized use or disclosure of the Company’s confidential information or trade secrets or any successor or affiliate of the Company that has, or may reasonably be expected to have, a material adverse impact on any such entity, (4) gross negligence, failure to follow a material, lawful and reasonable request of the Company or material violation of any duty of loyalty to the Company or any successor or affiliate of the Company, or any other demonstrable material misconduct by Ms. Morris, (5) ongoing and repeated failure or refusal to perform or neglect of her duties as required by her employment agreement, which failure, refusal or neglect continues for 30 days following Ms. Morris’s receipt of written notice from the Company stating with specificity the nature of such failure, refusal or neglect, or (6) material breach of any Company policy or any material provision of the Morris Agreement.
For purposes of the Morris Agreement, “Good Reason” generally means (1) a material diminution in Ms. Morris’s title, authority, duties or responsibilities, (2) a material diminution in the executive officer’s base compensation, unless such a reduction is imposed across-the-board to the Company’s senior management and such reduction is not greater than 15%, (3) a material change in the geographic location at which the executive officer must perform her duties, (4) any other action or inaction that constitutes a material breach by the Company or any successor or affiliate of the Company’s obligations to Ms. Morris under the Agreement, or (5) the Company elects not to renew the Agreement for another term.
Directors Compensation Table
Each of our non-employee directors, other than the lead director, receives an annual cash retainer of $55,000; the retainer for the lead director is $75,000. In addition, during 2025, each of our non-employee directors received stock options to purchase shares of our common stock valued at $188,830 as determined by the Black Scholes method on the date of grant, which vest on the next annual meeting of stockholders. The following table sets forth summary information concerning the total compensation paid to our non-employee directors in 2025 for services to our Company.
| Name | Cash Compensation ($) | Option Awards ($)(1) | Total ($) | |||||||||
| Richard Bagger | $ | 55,000 | $ | 188,830 | $ | 243,830 | ||||||
| Margaret Smith Bell | $ | 55,000 | $ | 188,830 | $ | 243,830 | ||||||
| David Grange | $ | 55,000 | $ | 188,830 | $ | 243,830 | ||||||
| James Hunter | $ | 30,250 | $ | 249,957 | $ | 280,207 | ||||||
| Adeoye Olukotun | $ | 55,000 | $ | 188,830 | $ | 243,830 | ||||||
| Newcomb Stillwell | $ | 55,000 | $ | 188,830 | $ | 243,830 | ||||||
| Carolyn Taylor | $ | 55,000 | $ | 188,830 | $ | 243,830 | ||||||
| James Treco(2) | $ | 75,000 | $ | 188,830 | $ | 263,830 | ||||||
| Total: | $ | 435,250 | $ | 1,571,767 | $ | 2,007,017 | ||||||
| (1) | Represents the aggregate grant date fair value of stock options granted in accordance with FASB ASC Topic 718. For the relevant assumptions used in determining these amounts, refer to Note 14 to our audited financial statements. These amounts do not necessarily correspond to the actual value that may be recognized from the stock option grant. | |
| (2) | Mr. Treco received additional cash compensation for serving as lead director. |
As of December 31, 2025, our non-employee directors listed in the table above held outstanding stock options, as follows:
| Name | Number of Shares Underlying Outstanding Stock Options | |||
| Richard Bagger | 10,042 | |||
| Margaret Smith Bell | 10,045 | |||
| David Grange | 10,045 | |||
| James Hunter | 7,744 | |||
| Adeoye Olukotun | 10,044 | |||
| Newcomb Stillwell | 10,041 | |||
| Carolyn Taylor | 10,041 | |||
| James Treco | 10,045 | |||
116
ITEM 12 – SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The following table sets forth certain information regarding beneficial ownership of our common stock as of March 11, 2026:
| ● | by each person who is known by us to beneficially own more than 5% of our common stock; |
| ● | by each of our officers and directors; and |
| ● | by all of our officers and directors as a group. |
Unless otherwise indicated in the footnotes to the following table, each person named in the table has sole voting and investment power and that person’s address is c/o Tonix Pharmaceuticals Holding Corp., 200 Connell Drive, Suite 3100, Berkeley Heights, New Jersey 07922.
| NAME OF OWNER | TITLE OF CLASS | NUMBER
OF SHARES OWNED |
PERCENTAGE
OF COMMON STOCK (1) |
|||||||
| 5% Holders | ||||||||||
| The Vanguard Group | Common Stock | 673,605 | (2) | 5.0 | ||||||
| BlackRock, Inc. | Common Stock | 845,544 | (3) | 6.3 | ||||||
| Point72 Asset Management, L.P. | Common Stock | 1,235,058 | (4) | 9.2 | ||||||
| Directors and Executive Officers | ||||||||||
| Seth Lederman | Common Stock | 87,588 | (5) | * | ||||||
| Jessica Morris | Common Stock | 19,708 | (6) | * | ||||||
| Bradley Saenger | Common Stock | 17,357 | (7) | * | ||||||
| Gregory Sullivan | Common Stock | 19,543 | (8) | * | ||||||
| Siobhan Fogarty | Common Stock | 9,033 | (9) | * | ||||||
| Richard Bagger | Common Stock | 42 | (10) | * | ||||||
| Margaret Smith Bell | Common Stock | 45 | (11) | * | ||||||
| David Grange | Common Stock | 45 | (12) | * | ||||||
| James Hunter | Common Stock | 2 | (13) | * | ||||||
| Adeoye Olukotun | Common Stock | 43 | (14) | * | ||||||
| Newcomb Stillwell | Common Stock | 41 | (15) | * | ||||||
| Carolyn Taylor | Common Stock | 459 | (16) | * | ||||||
| James Treco | Common Stock | 294 | (17) | * | ||||||
| Officers and Directors as a Group (11 persons) | Common Stock | 154,200 | (18) | * | % | |||||
* Denotes less than 1%
(1) Percentage based upon 13,405,401 shares of common stock issued and outstanding as of March 11, 2026.
(2) Based solely on Amendment No. 1 to the stockholder’s Schedule 13G filed on January 20, 2026. The stockholder has shared voting power with respect to 79,137 shares of common stock and shared dispositive power with respect to 673,605 shares of common stock. The address of the stockholder is 100 Vanguard Blvd., Malvern, PA 19355.
(3) Based solely on Amendment No. 1 to the stockholder’s Schedule 13G filed on January 20, 2026. The stockholder has sole voting and dispositive power with respect to 845,544.00 shares of common stock. The address of the stockholder is 50 Hudson Yards New York, NY 10001.
(4) Based solely on the Schedule 13G filed by Point72 Asset Management, L.P., Point72 Capital Advisors, Inc., and Steven A. Cohen on December 31, 2025. The address of the stockholder is 72 Cummings Point Road, Stamford, CT 06902. Includes 615,025 shares of common stock issuable upon exercise of warrants.
(5) Includes 83,581 shares of common stock underlying options which are currently exercisable or become exercisable within 60 days, 1 share of common stock owned by Lederman & Co, and 1 share owned through an IRA account. Seth Lederman, as the Managing Member of Lederman & Co has investment and voting control over the shares held by these entities.
(6) Includes 19,708 shares of common stock underlying options which are currently exercisable or become exercisable within 60 days.
(7) Includes 17,357 shares of common stock underlying options which are currently exercisable or become exercisable within 60 days.
(8) Includes 19,543 shares of common stock underlying options which are currently exercisable or become exercisable within 60 days.
(9) Includes 9,033 shares of common stock underlying options and restricted stock units which are currently exercisable or vested or become exercisable within 60 days.
(10) Includes 42 shares of common stock underlying options and restricted stock units which are currently exercisable or vested or become exercisable within 60 days.
(11) Includes 45 shares of common stock underlying options and restricted stock units which are currently exercisable or vested or become exercisable within 60 days.
(12) Includes 45 shares of common stock underlying options and restricted stock units which are currently exercisable or vested or become exercisable within 60 days.
(13) Includes 2 shares of common stock underlying options and restricted stock units which are currently exercisable or vested or become exercisable within 60 days.
(14) Includes 43 shares of common stock underlying options which are currently exercisable or become exercisable within 60 days
(15) Includes 41 shares of common stock underlying options which are currently exercisable or become exercisable within 60 days
(16) Includes 41 shares of common stock underlying options which are currently exercisable or become exercisable within 60 days
(17) Includes 44 shares of common stock underlying options which are currently exercisable or become exercisable within 60 days
(18) Includes 149,525 shares of common stock underlying options which are currently exercisable or vested or become exercisable within 60 days, 1 share of common stock owned by Lederman & Co, and 1 share owned through an IRA account of Dr. Lederman.
117
ITEM 13 – CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
We have adopted a written related-person transactions policy that sets forth our policies and procedures regarding the identification, review, consideration and oversight of “related-party transactions.” For purposes of our policy only, a “related-party transaction” is a transaction, arrangement or relationship (or any series of similar transactions, arrangements or relationships) in which we and any “related party” are participants involving an amount that exceeds the lesser of $120,000 or one percent of the average of our total assets at year end for the last two completed fiscal years.
A related party is any executive officer, director or a holder of more than five percent of our common stock, including any of their immediate family members and any entity owned or controlled by such persons.
Where a transaction has been identified as a related-party transaction, our Chief Compliance Officer must present information regarding the proposed related-party transaction to our Audit Committee and Nominating and Corporate Governance Committee for review. The presentation must include a description of, among other things, the material facts, the direct and indirect interests of the related parties, the benefits of the transaction to us and whether any alternative transactions are available. To identify related-party transactions in advance, we rely on information supplied by our executive officers, directors and certain significant stockholders. In considering related-party transactions, our Nominating and Corporate Governance Committee will take into account the relevant available facts and circumstances including, but not limited to:
| ● | whether the transaction was undertaken in the ordinary course of our business; |
| ● | whether the related party transaction was initiated by us or the related party; |
| ● | whether the transaction with the related party is proposed to be, or was, entered into on terms no less favorable to us than terms that could have been reached with an unrelated third party; |
| ● | the purpose of, and the potential benefits to us from the related party transaction; |
| ● | the approximate dollar value of the amount involved in the related party transaction, particularly as it relates to the related party; |
| ● | the related party’s interest in the related party transaction, and |
| ● | any other information regarding the related party transaction or the related party that would be material to investors in light of the circumstances of the particular transaction. |
The Nominating and Corporate Governance Committee shall then make a recommendation to the Board, who will determine whether or not to approve of the related party transaction, and if so, upon what terms and conditions. In the event a director has an interest in the proposed transaction, the director must recuse himself or herself from the deliberations and approval.
Other than as set forth below, during the last two fiscal years, there have been no related party transactions.
December 2025 Registered Direct Offering
On December 29, 2025, we entered into a securities purchase agreement with an affiliate of Point72 Asset Management, L.P., a holder of 5% or more of our common stock, pursuant to which we sold 615,025 shares of common stock and pre-funded warrants to purchase up to 615,025 shares of common stock. The offering price per share of common stock was $16.26, and the offering price per share of pre-funded warrant was $16.259.
Board Independence
The Board has determined that (i) because Seth Lederman is an executive officer of the Company, he has a relationship which, in the opinion of the Board, would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and is not an “independent director” as defined in the Marketplace Rules of The NASDAQ Stock Market, (ii) because James Hunter was a former employee of the Company, he has a relationship which, in the opinion of the Board, would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and is not an “independent director” as defined in the Marketplace Rules of The NASDAQ Stock Market, and (iii) Richard Bagger, Margaret Smith Bell, David Grange, Adeoye Olukotun, Newcomb Stillwell, Carolyn Taylor and James Treco are each an independent director as defined in the Marketplace Rules of The NASDAQ Stock Market.
118
ITEM 14 – PRINCIPAL ACCOUNTING FEES AND SERVICES
Our
independent registered public accounting firm is
(1) Audit Fees
The aggregate fees billed by our independent registered public accounting firm, for professional services rendered for the audit of our annual financial statements for the years ended December 31, 2025 and 2024, including review of our interim financial statements as well as registration statement filings with the SEC and comfort letters issued to underwriters were $537,075 and $648,375, respectively.
(2) Audit-Related Fees
We did not incur fees to our independent registered public accounting firm for audit related fees during the fiscal years ended December 31, 2025 and 2024.
(3) Tax Fees
We did not incur fees to our independent registered public accounting firm for tax services during the fiscal years ended December 31, 2025 and 2024.
(4) All Other Fees
None.
Pre-Approval Policies and Procedures
Consistent with SEC policies and guidelines regarding audit independence, the Audit Committee is responsible for the pre-approval of all audit and permissible non-audit services provided by our principal accountants on a case-by-case basis. Our Audit Committee has established a policy regarding approval of all audit and permissible non-audit services provided by our principal accountants. Our Audit Committee pre-approves these services by category and service. Our Audit Committee has pre-approved all of the services provided by our principal accountants.
119
PART IV
ITEM 15 – EXHIBITS, FINANCIAL STATEMENT SCHEDULES
| (c) | Index to Exhibits |
The Exhibits listed below are identified by numbers corresponding to the Exhibit Table of Item 601 of Regulation S-K. The Exhibits designated by an asterisk (*) are management contracts or compensatory plans or arrangements required to be filed pursuant to Item 15.
EXHIBIT INDEX
| Exhibit
No. |
Description |
| 3.01 | Articles of Incorporation, filed as an exhibit to the Registration Statement on Form S-1, filed with the Securities and Exchange Commission (the “Commission”) on April 9, 2008 and incorporated herein by reference. |
| 3.02 | Articles of Merger between Tamandare Explorations Inc. and Tonix Pharmaceuticals Holding Corp., effective October 11, 2011, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 17, 2011 and incorporated herein by reference. |
| 3.03 | Third Amended and Restated Bylaws, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on June 3, 2016 and incorporated herein by reference. |
| 3.04 | Certificate of Change of Tonix Pharmaceuticals Holding Corp., dated March 13, 2017 and effective March 17, 2017, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on March 16, 2017 and incorporated herein by reference. |
| 3.05 | Certificate of Amendment to Articles of Incorporation, effective June 16, 2017, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on June 16, 2017 and incorporated herein by reference. |
| 3.06 | Certificate of Amendment to Tonix Pharmaceuticals Holding Corp.’s Articles of Incorporation, as amended, filed with the Secretary of State of the State of Nevada on May 3, 2019. |
| 3.07 | Form of Certificate of Designation of Series A Convertible Preferred Stock, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 25, 2022 and incorporated herein by reference. |
| 3.08 | Form of Certificate of Designation of Series B Convertible Preferred Stock, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on October 25, 2022 and incorporated herein by reference. |
| 3.09 | Certificate of Amendment to Tonix Pharmaceuticals Holding Corp.’s Articles of Incorporation, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on May 16, 2022 and incorporated herein by reference. |
| 3.10 | Certificate of Amendment to Tonix Pharmaceuticals Holding Corp.’s Articles of Incorporation, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on January 25, 2024 and incorporated herein by reference |
| 4.01 | Specimen Common Stock Certificate of the Registrant, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on May 24, 2018 and incorporated herein by reference. |
| 4.02 | Description of Registrant’s Securities, filed herewith. |
| 4.03 | Form of Common Warrant, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on July 28, 2023 and incorporated herein by reference. |
| 4.04 | Form of Series C Warrant. filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on December 21, 2023 and incorporated herein by reference. |
| 4.05 | Form of Series D Warrant. filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on December 21, 2023 and incorporated herein by reference. |
| 4.06 | Form of Series E Warrant, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on March 29, 2024, and incorporated herein by reference. |
| 4.07 | Amendment to Common Stock Purchase Warrant, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on March 29, 2024, and incorporated herein by reference. |
| 4.08 | Form of Pre-Funded Warrant, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on December 29, 2025 and incorporated herein by reference. |
120
| 10.01 | Employment Agreement, between Tonix Pharmaceuticals Holding Corp. and Seth Lederman, dated February 11, 2014, filed as an exhibit to the Current Report on Form 8-K filed with the Commission on February 14, 2014 and incorporated herein by reference.* |
| 10.02 | Tonix Pharmaceuticals Holding Corp. 2014 Stock Incentive Plan, incorporated herein by reference to Annex A to our Definitive Proxy Statement on Schedule 14A (File No. 001-36019), filed with the Commission on May 2, 2014.* |
| 10.03 | Lease Amendment and Expansion Agreement, dated February 11, 2014, by and between 509 Madison Avenue Associates, L.P. and Tonix Pharmaceuticals, Inc., filed as an exhibit to the Annual Report on Form 10-K filed with the Commission on February 27, 2015 and incorporated herein by reference. |
| 10.04 | Employment Agreement, between Tonix Pharmaceuticals Holding Corp. and Gregory Sullivan, dated June 3, 2014, filed as an exhibit to the Current Report on Form 8-K filed with the Commission on June 3, 2014 and incorporated herein by reference.* |
| 10.05 | Tonix Pharmaceuticals Holding Corp. 2016 Stock Incentive Plan, incorporated herein by reference to Annex A to our Definitive Proxy Statement on Schedule 14A (File No. 001-36019), filed with the Commission on March 25, 2016.* |
| 10.06 | Tonix Pharmaceuticals Holding Corp. 2017 Stock Incentive Plan, incorporated herein by reference to Appendix A to our Definitive Proxy Statement on Schedule 14A (File No. 001-36019), filed with the Commission on May 2, 2017.* |
| 10.07 | Tonix Pharmaceuticals Holding Corp. 2018 Equity Incentive Plan, incorporated herein by reference to our Definitive Proxy Statement on Schedule 14A (File No. 001-36019), filed with the Commission on April 19, 2018.* |
| 10.08 | Purchase Agreement, dated October 18, 2018, between Tonix Pharmaceuticals Holding Corp. and Lincoln Park Capital Fund, LLC, filed as an exhibit to the Current Report on Form 8-K filed with the Commission on October 24, 2018 and incorporated herein by reference. |
| 10.09 | Tonix Pharmaceuticals Holding Corp. 2019 Stock Incentive Plan, incorporated herein by reference to Appendix A to our Definitive Proxy Statement on Schedule 14A (File No. 001-36019), filed with the Commission on March 18, 2019.* |
| 10.10 | Tonix Pharmaceuticals Holding Corp. 2019 Employee Stock Purchase Plan, incorporated herein by reference to Appendix B to our Definitive Proxy Statement on Schedule 14A (File No. 001-36019), filed with the Commission on March 18, 2019.* |
| 10.11 | License Agreement, dated May 20, 2019, between Tonix Pharmaceuticals Holding Corp. and The Trustees of Columbia University in the City of New York, filed as an exhibit to the Quarterly Report on Form 10-Q filed with the Commission on August 12, 2019 and incorporated herein by reference. |
121
| 10.12 | Asset Purchase Agreement, dated August 19, 2019, between Tonix Pharmaceuticals Holding Corp. and TRImaran Pharma, Inc., filed as an exhibit to the Quarterly Report on Form 10-Q filed with the Commission on November 8, 2019 and incorporated herein by reference. |
| 10.13 | First Amended and Restated Exclusive License Agreement, dated August 19, 2019, between Tonix Pharmaceuticals Holding Corp. and Wayne State University, filed as an exhibit to the Quarterly Report on Form 10-Q filed with the Commission on November 8, 2019 and incorporated herein by reference. |
| 10.14 | Exclusive License Agreement, dated September 16, 2019, between Tonix Pharmaceuticals Holding Corp. and The Trustees of Columbia University in the City of New York, filed as an exhibit to the Quarterly Report on Form 10-Q filed with the Commission on November 8, 2019 and incorporated herein by reference. |
| 10.15 | Tonix Pharmaceuticals Holding Corp. 2020 Stock Incentive Plan, incorporated herein by reference to Appendix A to our Definitive Proxy Statement on Schedule 14A (File No. 001-36019), filed with the Commission on December 13, 2019.* |
| 10.16 | Research Collaboration Agreement between Tonix Pharmaceutical, Inc. and Southern Research Institute, dated November 7, 2018, filed as an exhibit to the Annual Report on Form 10-K, filed with the Commission on March 24, 2020 and incorporated herein by reference. |
| 10.17 | License Agreement, dated May 5, 2020, between Tonix Pharmaceuticals (Canada) Inc. and The Governors of the University of Alberta, filed as an exhibit to the Quarterly Report on Form 10-Q, filed with the Commission on August 10, 2020 and incorporated herein by reference.† |
| 10.18 | Asset Purchase Agreement, dated June 11, 2020, between Tonix Pharmaceuticals, Inc. and Trigemina, Inc., filed as an exhibit to the Quarterly Report on Form 10-Q, filed with the Commission on May 12, 2020 and incorporated herein by reference.† |
| 10.19 | Amended and Restated Exclusive License Agreement, dated June 11, 2020, between Tonix Pharmaceuticals, Inc. and The Board of Trustees of the Leland Stanford Junior University, filed as an exhibit to the Quarterly Report on Form 10-Q, filed with the Commission on August 10, 2020 and incorporated herein by reference. |
| 10.20 | Assignment and Agreement, dated June 11, 2020, between Tonix Pharmaceuticals, Inc. and The Board of Trustees of the Leland Stanford Junior University, filed as an exhibit to the Quarterly Report on Form 10-Q, filed with the Commission on August 10, 2020 and incorporated herein by reference. |
| 10.21 | Tonix Pharmaceuticals Holding Corp. Amended and Restated 2020 Stock Incentive Plan, incorporated herein by reference to Appendix A to our Definitive Proxy Statement on Schedule 14A (File No. 001-36019), filed with the Commission on March 30, 2020.* |
122
| 10.22 | Employment Agreement, between Tonix Pharmaceuticals Holding Corp. and Jessica Morris, dated February 23, 2021, filed as an exhibit to the Current Report on Form 8-K filed with the Commission on February 26, 2021 and incorporated herein by reference.* |
| 10.23 | Employment Agreement, between Tonix Pharmaceuticals Holding Corp. and Bradley Saenger, dated February 23, 2021, filed as an exhibit to the Current Report on Form 8-K filed with the Commission on February 26, 2021 and incorporated herein by reference.* |
| 10.24 | License Agreement, dated April 14, 2021, between the Company and OyaGen, Inc., filed as an exhibit to the Quarterly Report on Form 10-Q filed with the Commission on May 10, 2021 and incorporated herein by reference.† |
| 10.25 | Purchase Agreement, dated August 16, 2022, by and between Tonix Pharmaceuticals Holding Corp. and Lincoln Park Capital Fund, LLC, filed as an exhibit to the Current Report on Form 8-K filed with the Commission on August 17, 2022, and incorporated herein by reference. |
| 10.26 | Tonix Pharmaceuticals Holding Corp. 2022 Employee Stock Purchase Plan, incorporated herein by reference to Appendix A to our Definitive Proxy Statement on Schedule 14A, filed with the Commission on March 18, 2022.* |
| 10.27 | Asset Purchase Agreement, dated as of June 23, 2023, by and among Upsher-Smith Laboratories, LLC, Tonix Medicines, Inc. and Tonix Pharmaceuticals Holding Corp., filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on June 26, 2023, and incorporated herein by reference. |
| 10.28 | Transition Services Agreement, dated as of June 30, 2023, by and among Upsher-Smith Laboratories, LLC and Tonix Medicines Inc., filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on July 3, 2023, and incorporated herein by reference. |
123
| 10.29 | Loan and Guaranty Agreement, dated as of December 8, 2023, by and among the Loan Parties, the Lenders and the JGB Agent, filed with the Commission on December 8, 2023, and incorporated herein by reference. |
| 10.30 | Placement Agent Agreement, dated March 28, 2024, between Tonix Pharmaceuticals Holding Corp. and A.G.P./Alliance Global Partners, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on March 29, 2024 and incorporated herein by reference. |
| 10.31 | Form of Series C Warrant. filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on December 21, 2023 and incorporated herein by reference. |
| 10.32 | Form of Series D Warrant. filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on December 21, 2023 and incorporated herein by reference. |
| 10.33 | Placement Agent Agreement, dated March 28, 2024, between Tonix Pharmaceuticals Holding Corp. and A.G.P./Alliance Global Partners, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on March 29, 2024 and incorporated herein by reference. |
| 10.34 | Placement Agency Agreement, dated June 12, 2024, between Tonix Pharmaceuticals Holding Corp. and Dawson James Securities Inc., filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on June 13, 2024, and incorporated herein by reference. |
| 10.35 | Warrant Agent Agreement, dated June 13, 2024, between Tonix Pharmaceuticals Holding Corp. and VStock Transfer, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on June 13, 2024 and incorporated herein by reference. |
| 10.36 | Placement Agency Agreement, dated June 27, 2024, between Tonix Pharmaceuticals Holding Corp. and Dawson James Securities Inc., filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on June 28, 2024 and incorporated herein by reference. |
| 10.37 | Warrant Agent Agreement, dated June 28, 2024, between Tonix Pharmaceuticals Holding Corp. and VStock Transfer, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on June 28, 2024 and incorporated herein by reference. |
| 10.38 | Placement Agency Agreement, dated July 9, 2024, between Tonix Pharmaceuticals Holding Corp. and Dawson James Securities Inc., filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on July 10, 2024 and incorporated herein by reference. |
| 10.39 | Warrant Agent Agreement, dated July 10, 2024, between Tonix Pharmaceuticals Holding Corp. and VStock Transfer, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on July 10, 2024 and incorporated herein by reference. |
| 10.40 | Form of Security Purchase Agreement, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on July 10, 2024 and incorporated herein by reference. |
| 10.41 | Sales Agreement, dated July 30, 2024, between Tonix Pharmaceuticals Holding Corp. and A.G.P./ Alliance Global Partners, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on July 30, 2024 and incorporated herein by reference. |
124
| 10.42 | Pay-Off Letter, dated February 3, 2025, by and among the Loan Parties, the Lenders and the JGB Agent, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on February 3, 2025 and incorporated herein by reference.† |
| 10.43 | Base Agreement between Advanced Technology International (ATI) and Tonix Pharmaceuticals, Inc., dated July 17, 2023, filed as an exhibit to the Annual Report on Form 10-K, filed with the Commission on March 18, 2025. |
| 10.44 | Project Agreement No. 1 by and between Advanced Technology International and Tonix Pharmaceuticals, Inc., dated June 28, 2024, filed as an exhibit to the Annual Report on Form 10-K, filed with the Commission on March 18, 2025.† |
| 10.45 |
Sales Agreement, dated June 11, 2025, by and between Tonix Pharmaceuticals Holdings Corp. and A.G.P./Alliance Global Partners, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on June 11, 2025 and incorporated herein by reference. |
| 10.46 |
Purchase Agreement, dated June 11, 2025, by and between Tonix Pharmaceuticals Holding Corp. and Lincoln Park Capital Fund, LLC, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on June 11, 2025 and incorporated herein by reference. † |
| 10.47 |
Registration Rights Agreement, dated June 11, 2025, by and between Tonix Pharmaceuticals Holding Corp. and Lincoln Park Capital Fund, LLC, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on June 11, 2025 and incorporated herein by reference. † |
| 10.48 | Amendment No. 1 to Sales Agreement, dated November 21, 2025, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on November 21, 2025 and incorporated herein by reference. |
| 10.49 |
Placement Agency Agreement, dated December 29, 2025, between Tonix Pharmaceuticals Holding Corp. and TD Securities (USA) LLC, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on December 29, 2025 and incorporated herein by reference. |
| 10.50 | Form of Securities Purchase Agreement, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on December 29, 2025 and incorporated herein by reference. |
| 10.51 | Exclusive License Agreement, dated June 26, 2025, by and between University of Massachusetts and Tonix Pharmaceuticals, Inc., filed herewith † |
| 14.01 | Code of Business Conduct and Ethics for Employees, Executive Officers and Directors, filed as an exhibit to the Current Report on Form 8-K, filed with the Commission on February 16, 2016, and incorporated herein by reference. |
| 19.01 | Tonix Pharmaceuticals Holding Corp. Insider Trading Policy, filed as an exhibit to the Annual Report on Form 10-K, filed with the Commission on March 18, 2025. |
| 21.01 | List of Subsidiaries, filed herewith. |
| 23.01 | Consent of Independent Registered Public Accounting Firm, filed herewith. |
| 31.01 | Certification of Chief Executive Officer pursuant to Exchange Act Rules 13a-14(a) and 15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002, filed herewith. |
| 31.02 | Certification of Chief Financial Officer pursuant to Exchange Act Rules 13a-14(a) and 15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002, filed herewith. |
| 32.01 | Certifications of Chief Executive Officer and Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, filed herewith. |
| 97.01 | Tonix Pharmaceuticals Holding Corp. Compensation Recovery Policy, filed as an exhibit to the Annual Report on Form 10-K, filed with the Commission on March 18, 2025. |
| 101 | The following materials from Tonix Pharmaceuticals Holding Corp.’s Annual Report on Form 10-K for the year ended December 31, 2023, formatted in XBRL (Extensible Business Reporting Language): (i) the Consolidated Balance Sheets, (ii) the Consolidated Statements of Operations, (iii) the Consolidated Statements of Comprehensive Loss, (iv) the Consolidated Statements of Stockholders’ Equity, (v) the Consolidated Statements of Cash Flows, and (vi) Notes to Consolidated Financial Statements. |
| 104 | The cover page from this Annual Report on Form 10-K, formatted as Inline XBRL. |
| † | Certain portions of this exhibit, that are not material and would likely cause competitive harm to the registrant if publicly disclosed, have been redacted pursuant to Item 601(b)(10) of Regulation S-K. |
| †† | This certification is not deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, or otherwise subject to the liability of that section. Such certification will not be deemed to be incorporated by reference into any filing under the Securities Act of 1933 or the Securities Exchange Act of 1934, except to the extent that the registrant specifically incorporates it by reference. |
| * | Denotes a management compensatory agreement or arrangement. |
125
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| TONIX PHARMACEUTICALS HOLDING CORP. | ||
| Date: March 12, 2026 | By: | /s/ SETH LEDERMAN |
| Seth Lederman | ||
| Chief Executive Officer (Principal Executive Officer) | ||
| Date: March 12, 2026 | By: | /s/ BRADLEY SAENGER |
| Bradley Saenger | ||
| Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) | ||
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Seth Lederman and Bradley Saenger, jointly and severally, his or her attorney-in-fact, with the power of substitution, for him or her in any and all capacities, to sign any amendments to this Annual Report on Form 10-K and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each of said attorneys-in-fact, or his or her substitute or substitutes, may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Annual Report on Form 10-K has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| Name | Position | Date | ||
| /s/ SETH LEDERMAN | Chief Executive Officer, President and Director (Principal Executive Officer) | March 12, 2026 | ||
| Seth Lederman | ||||
| /s/ BRADLEY SAENGER | Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) | March 12, 2026 | ||
| Bradley Saenger | ||||
| /s/ RICHARD BAGGER | Director | March 12, 2026 | ||
| Richard Bagger | ||||
| /s/ MARGARET SMITH BELL | Director | March 12, 2026 | ||
| Margaret Smith Bell | ||||
| /s/ DAVID GRANGE | Director | March 12, 2026 | ||
| David Grange | ||||
| /s/ JAMES HUNTER | Director | March 12, 2026 | ||
James Hunter
|
||||
| /s/ ADEOYE OLUKOTUN | Director | March 12, 2026 | ||
Adeoye Olukotun
|
||||
| /s/ NEWCOMB STILLWELL | Director | March 12, 2026 | ||
| Newcomb Stillwell | ||||
| /s/ CAROLYN TAYLOR | Director | March 12, 2026 | ||
Carolyn Taylor
|
||||
| /s/ JAMES TRECO | Director | March 12, 2026 | ||
| James Treco |
126